

Warum erwarten wir das Schlimmste in den kommenden 5 - 9 Jahren?
miR, KRAS und der ganze Horror der Transfektionsschüsse - Ein MTV-Ausschnitt für Fortgeschrittene, was in einer eukaryotischen Zelle so abgeht.


“Als Wissenschaftler glaube ich nichts. Die Wissenschaft sollte das Wort Glauben nicht verwenden. Es gibt Dinge, die wahrscheinlicher und weniger wahrscheinlich sind. Die Wissenschaft kann nichts mit absoluter Gewissheit sagen." - Lawrence M. Krauss
https://edition.cnn.com/2022/10/14/health/early-onset-cancer-increase/index.html
“Es könnte sich eine weltweite Krebsepidemie bei Menschen unter 50 Jahren entwickeln”
Aus gegebenem Anlass werde ich mich wohl oder übel meinem Hass-Liebe-Thema: dem onkogene Prozesse auslösendem KRAS widmen müssen.
Wo fängt man an, wenn man schon halb im Thema Epigenetik gelandet ist, will man über micro RNAs (miR) und Genregulation reden? Vielleicht damit, zu erwähnen, dass der in diesem Substack diskutierte Weg, sich mal richtig hübsch ein krebsregulatorisches Gen, wie das p53, zu versauen, wenn man sich den goldenen Pikser holt, nur einer von vielen versteckten Mechanismen wäre. Des Weiteren muss ich wohl zunächst erneut darauf verweisen, kein studierter Fachmann auf diesem Gebiet zu sein und mir alles, was ich hier diskutieren will selbst beibringe, in dem ich anderen, die viel mehr Ahnung haben, zuhöre und ihnen Löcher in den Bauch frage. Somit sind Lücken und eventuelle Denkfehler natürlich nicht auszuklammern. Dann muss ich noch anmerken, dass allein die Regulierung der einzelnen Zellen und deren miRs schon eine extrem entscheidende Rolle für die weiteren Prozesse nach der “was-auch-immer-das-ist”-RNA-Pikse spielen werden, genauso wie die unterschiedlichen Chargen, deren RNA-Integrität und persönliche epigenetische Faktoren jedes Menschen.
Bevor es losgeht: Ich werde auch wieder im Laufe dieses Artikels auf einige meiner Substacks verweisen, da es meine persönlichen Lernprozesse sind, die mich jetzt zu diesem Artikel führten.
Doch wo starte ich nun. Das ist ja die eigentliche Frage, da wir - wenn wir von Genen reden - schon fast am Ende der Transfektionsprozesse angekommen sind.
Ich denke, ich werde mit diesem Paper anfangen, da hier gut sichtbar gemacht wird, was so alles schief gehen kann, wenn man das KRAS hochreguliert:
https://www.lungcancerjournal.info/article/S0169-5002(18)30473-2/fulltext
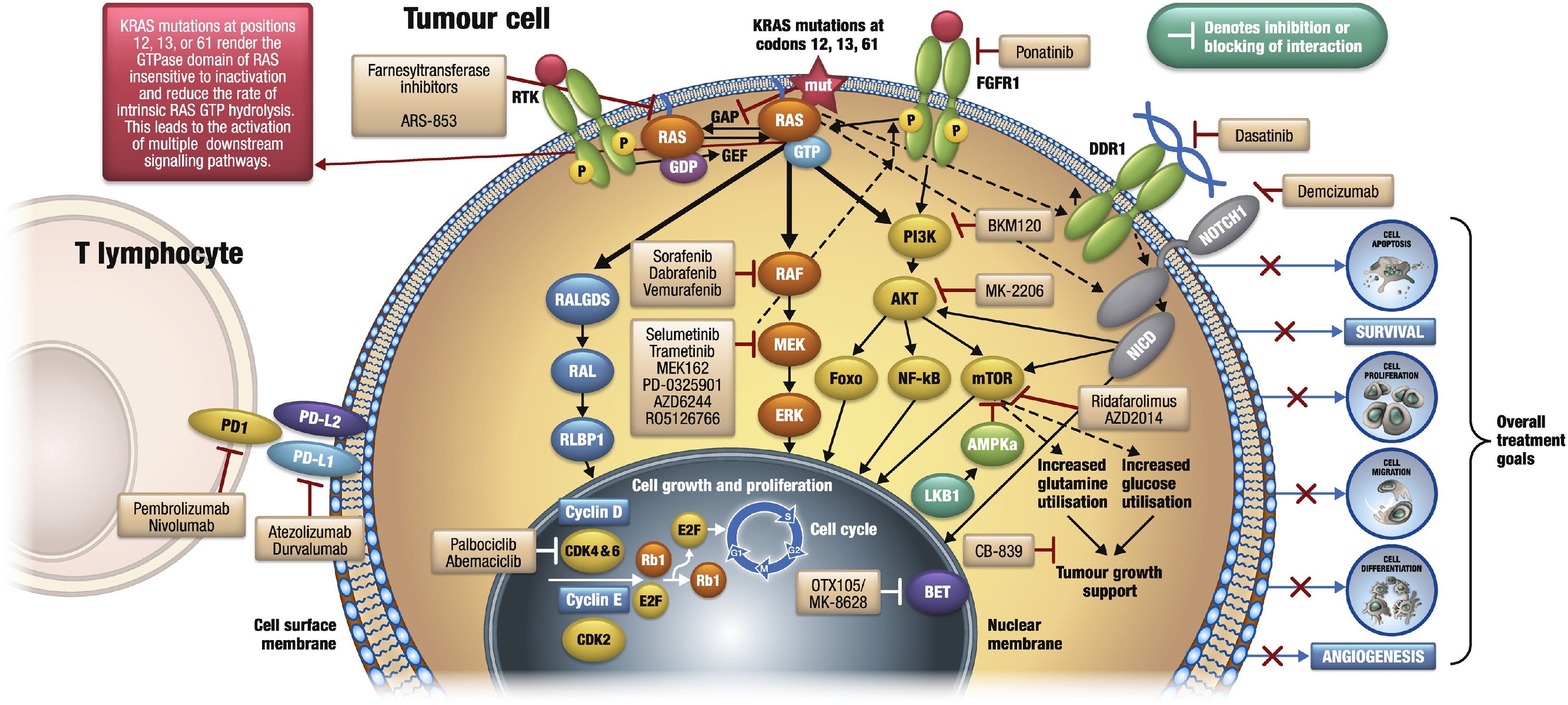
“2.1 KRAS-Funktion und wichtige nachgeschaltete Effektorwege
KRAS, ein Mitglied der menschlichen RAS-Genfamilie, kodiert für ein membrangebundenes kleines GTPase-Protein, das in zwei verschiedenen Zuständen vorliegen kann: GDP-gebunden, was inaktiv ist, und GTP-gebunden, was aktiv ist und Signale durch Interaktion mit verschiedenen nachgeschalteten Effektoren überträgt. RAS-Proteine fungieren als zellulärer Schalter, der durch extrazelluläre Reize eingeschaltet wird, was zur vorübergehenden Bildung der aktiven, GTP-gebundenen Form von RAS führt, die verschiedene Signalwege aktiviert, die grundlegende Zellprozesse regulieren. Die Aktivierung des RAS-Signalwegs wird durch Regulatorfaktoren reguliert, die den GDP-GTP-Austausch bewirken (Guanin-Nukleotid-Austauschfaktoren; GEFs) oder seine GTPase-Aktivität beeinflussen (GTPase-aktivierende Proteine; GAPs). GEFs erhöhen die Freisetzung von GDP aus RAS und ermöglichen dessen Austausch gegen GTP, was zur Aktivierung von RAS führt. GAPs erhöhen die intrinsische GTPase-Aktivität von RAS, was zu einer schnellen Umwandlung von aktivem zu inaktivem GDP-gebundenem RAS führt. Mutierte RAS-Onkoproteine unterscheiden sich funktionell von ihren normalen Gegenstücken dadurch, dass die onkogenen Formen GAP daran hindern, die intrinsische katalytische Rate der GTPase zu erhöhen, wodurch RAS in seinem konstitutiv GTP-gebundenen aktiven Zustand gehalten wird, der onkogene Stoffwechselwege und die zelluläre Signaltransduktion aktiviert.
Zusätzlich zur GTP-Bindung müssen RAS-Proteine mit Zellmembranen assoziieren, wo sie mit GEFs und anderen vorgeschalteten Regulatoren interagieren, um extrazelluläre Signale stromabwärts zu übertragen. RAS-Proteine werden als lösliche Vorstufen synthetisiert, die dann posttranslational modifiziert werden müssen, um ihre Assoziation mit Membranen zu vermitteln. Die Enzyme, die diese Modifikationen katalysieren, sind ebenfalls interessante Ziele für die Entwicklung von Anti-RAS-Therapien.
Das Signalnetzwerk, das RAS reguliert, bestimmt die biologischen Wirkungen von RAS. Aus diesem Grund ist es nicht nur wichtig zu verstehen, wie RAS aktiviert wird, sondern auch seine nachgeschalteten molekularen Effektoren zu kennen. RAS-Proteine spielen eine zentrale Rolle bei der Weiterleitung mitogener Signale, so dass ihre wichtigsten nachgeschalteten Effektoren an der Aktivierung mitogener Signalwege beteiligt sind, die in die Tumorentstehung involviert sind. Es gibt mehr als zehn bekannte RAS-Effektoren, von denen wir im Folgenden diejenigen beschreiben, die bei der RAS-vermittelten Tumorentstehung von größerer Bedeutung sind.”
Ich wollte jetzt nicht noch die einzelnen Effektoren beleuchten, da es auch so schon ein Höllenritt wird, was ich hier noch diskutieren will.
Warum wurde mRNA Technologie eigentlich gleich wieder vorrangig in der Krebsforschung als experimentelles Therapeutikum genutzt? Hm…. Ich sehe schon, wie die Mützen- und Perückenproduktion demnächst angekurbelt wird.
Und das mit den RAS-Effektoren etwas geschehen sein muss, schaut man sich im Supplementary 3 der folgenden Studie die Genregulation an, dürfte wohl offensichtlich sein:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9029844/

Sie wussten also bereits, dass es das KRAS-Gen hochregulieren würde mit spätestens dieser Publikation.
https://clinicaltrials.gov/ct2/show/NCT04895982
Ich würde euch ja nun an dieser Stelle gerne weitere Publikationen dazu präsentieren, ob es viele Menschen betreffen wird und wie wahrscheinlich Krebsentwicklungen - daraus resultierend - wären. Doch dazu fehlt ja - Witz komm raus - jedes halbwegs nachvollziehbare Datengerüst.
Also werde ich eine logische Kausalkette aufbauen, um zu zeigen, wieso es meiner bescheidenen Meinung nach realistisch erscheint und auch, wieso ein hochreguliertes KRAS nichts Gutes erahnen lässt.

https://en.wikipedia.org/wiki/KRAS
Doch Eins nach dem andern. Definieren wir doch erstmal zum besseren Verständnis, was genau wir nun eigentlich unter dem KRAS-Gen verstehen:
“KRAS (Kirsten-Ratten-Sarkom-Virus) ist ein Gen, das Anweisungen für die Herstellung eines Proteins namens K-Ras liefert, das Teil des RAS/MAPK-Wegs ist. Das Protein leitet Signale von außerhalb der Zelle an den Zellkern weiter. Diese Signale weisen die Zelle an, zu wachsen und sich zu teilen (Proliferation) oder zu reifen und spezialisierte Funktionen zu übernehmen (Differenzierung). Es heißt KRAS, weil es erstmals als virales Onkogen im Kirsten-Ras-Sarkom-Virus identifiziert wurde. Das identifizierte Onkogen stammte aus einem zellulären Genom, so dass KRAS, wenn es in einem zellulären Genom gefunden wird, als Proto-Onkogen bezeichnet wird.
Das K-Ras-Protein ist eine GTPase, eine Klasse von Enzymen, die das Nukleotid Guanosintriphosphat (GTP) in Guanosindiphosphat (GDP) umwandeln. Auf diese Weise wirkt das K-Ras-Protein wie ein Schalter, der von den GTP- und GDP-Molekülen ein- und ausgeschaltet wird. Um Signale zu übertragen, muss es durch Bindung an ein GTP-Molekül eingeschaltet werden. Das K-Ras-Protein wird ausgeschaltet (inaktiviert), wenn es das GTP in GDP umwandelt. Wenn das Protein an GDP gebunden ist, sendet es keine Signale an den Zellkern.
Das Genprodukt von KRAS, das K-Ras-Protein, wurde zuerst als p21-GTPase entdeckt. Wie andere Mitglieder der Ras-Unterfamilie der GTPasen ist das K-Ras-Protein ein früher Akteur in vielen Signaltransduktionswegen. K-Ras ist in der Regel an Zellmembranen gebunden, da es an seinem C-Terminus eine Isoprengruppe aufweist. In Säugetierzellen gibt es zwei Proteinprodukte des KRAS-Gens, die durch die Verwendung des alternativen Exons 4 (Exon 4A bzw. 4B) entstehen: K-Ras4A und K-Ras4B. Diese Proteine haben unterschiedliche Strukturen in ihrem C-terminalen Bereich und nutzen unterschiedliche Mechanismen zur Lokalisierung an Zellmembranen, einschließlich der Plasmamembran.”
Und jetzt bitte genaustens lesen und aufpassen:
”KRAS fungiert als molekularer Ein/Aus-Schalter, der die Proteindynamik nutzt. Sobald es allosterisch (‼ Allosterie ist die Eigenschaft vieler aus mehreren Untereinheiten zusammengesetzter Enzyme, ihre Raumstruktur unter der Beeinflussung des aktiven Bindungszentrums zu verändern. ‼) aktiviert ist, rekrutiert und aktiviert es Proteine, die für die Ausbreitung von Wachstumsfaktoren erforderlich sind, sowie andere Zellsignalrezeptoren wie c-Raf und PI-3-Kinase. KRAS reguliert den Glukosetransporter GLUT1 hoch und trägt damit zum Warburg-Effekt in Krebszellen bei. KRAS bindet in seinem aktiven Zustand an GTP. Es besitzt auch eine eigene enzymatische Aktivität, die das terminale Phosphat des Nukleotids spaltet und es in GDP umwandelt. Nach der Umwandlung von GTP in GDP wird KRAS deaktiviert. Die Umwandlungsrate ist in der Regel langsam, kann aber durch ein akzessorisches Protein der Klasse der GTPase-aktivierenden Proteine (GAP), z. B. RasGAP, drastisch erhöht werden. Im Gegenzug kann KRAS an Proteine der Klasse der Guanin-Nukleotid-Austauschfaktoren (GEF) binden (z. B. SOS1), was die Freisetzung des gebundenen Nukleotids (GDP) erzwingt. Anschließend bindet KRAS im Zytosol vorhandenes GTP und der GEF wird von ras-GTP befreit.
Andere Mitglieder der Ras-Familie sind: HRAS und NRAS. Diese Proteine werden alle auf die gleiche Weise reguliert und scheinen sich in ihren Wirkorten innerhalb der Zelle zu unterscheiden.”
Aufgepasst? KRAS reguliert also die Glukosetransporter hoch. Was wussten wir bereits über SC-2?!
https://www.degruyter.com/document/doi/10.1515/dmpt-2022-0148/html

“Dieses Phänomen ist auch als "Warburg-Effekt" bekannt und gilt hauptsächlich für Krebszellen und wurde für einige Viren überprüft. SARS-CoV-2 kann in den Zellen, die es infiziert, ebenfalls eine Warburg-ähnliche Aktivität zeigen, doch muss dies noch geklärt werden, da die aerobe Glykolyse in Alveolarepithelzellen unabhängig von der Virusinfektion als Reaktion auf Hypoxie aktiviert werden kann.”
Klären wir schnell den Begriff “Hypoxie”: Unter einer Hypoxie versteht man die Minderversorgung des Körpers oder einzelner Körperabschnitte mit Sauerstoff (O2).
Weiter heißt es dann:
”Metabolische Veränderungen in Immunzellen wurden ebenfalls in mehreren Studien untersucht. Eine der gut konzipierten Studien zeigte, dass SARS-CoV-2-infizierte Monozyten das Niveau der Glykolyse, die glykolytische Kapazität und die glykolytische Reserve, die für die Förderung der viralen Replikation erforderlich sind, erhöhten. Zur Untermauerung der metabolischen Veränderung von Zellen wurde in vielen anderen Studien auch festgestellt, dass SARS-CoV-2-infizierte oder betroffene Zellen ihre Stoffwechselfunktionen umstellen und schließlich nicht mehr funktionsfähig sind.”
Ich denke, wenn ihr bis hierher aufmerksam gefolgt seid, dürftet ihr eine langsame Vorstellung davon kriegen, wieso ich mich so auf dieses Gen versteift habe. Und wieso es mir einen kalten Schauer den Rücken runterlaufen lässt, dass dieses nach dem Schuss hochreguliert ist. Doch warum genau macht es mir nach dem Schuss soviel mehr Angst, als nach einer Infektion? Und hier wird es sehr kompliziert:
Signalkaskaden. Da war doch was? Dazu - solltet ihr mit diesem Begriff gar nichts anfangen können - müsstet ihr euch wohl oder übel noch einmal durch mein Substack über doppelgesichtige Götter durchackern:

Über den doppelgesichtigen Gott (Deutsch)
Über den doppelgesichtigen Gott (Englisch)
Wie wird das RAS-Protein nun also aktiviert und die Signalkaskade MAP-kinase (RAS-RAF-MEK-ERK)?
Das weiß sogar Wikipedia:
“Rezeptor-gebundene Tyrosinkinasen, wie der epidermale Wachstumsfaktor-Rezeptor (EGFR), werden durch extrazelluläre Liganden, wie den epidermalen Wachstumsfaktor (EGF), aktiviert. Die Bindung von EGF an den EGFR aktiviert die Tyrosinkinase-Aktivität der zytoplasmatischen Domäne des Rezeptors. Der EGFR wird an Tyrosinresten phosphoryliert. Docking-Proteine wie GRB2 enthalten eine SH2-Domäne, die an die Phosphotyrosinreste des aktivierten Rezeptors bindet. GRB2 bindet über die beiden SH3-Domänen von GRB2 an den Guanin-Nukleotid-Austauschfaktor SOS. Wenn der GRB2-SOS-Komplex an den phosphorylierten EGFR andockt, wird SOS aktiviert. Aktiviertes SOS fördert dann die Entfernung von GDP von einem Mitglied der Ras-Unterfamilie (vor allem H-Ras oder K-Ras). Das Ras-Protein kann dann GTP binden und aktiv werden.”
Und jetzt bitte anschnallen. Natürlich findet man nicht wirklich Papers, welche über die rezeptor-gebundene Tyrosinkinasen berichten möchten. Denn Vorsicht: Damit könnte man schon vorhersagen, dass nichts Gutes nach dem Schuss bei rauskommen wird, pfuscht man mal eben in diesen rum. Doch es gibt ein Preprint, welches Rückschlüsse zulässt. Denn die Autoren beschrieben, dass sie bei Krebspatienten mit Tyrosin-Inhibitoren eine schwächere Immunantwort beobachteten:
https://www.medrxiv.org/content/10.1101/2021.04.15.21255482v1.full

"Die Immunantwort auf eine Impfung kann tatsächlich durch Tyrosinkinase-Inhibitoren (TKI) abgeschwächt werden, insbesondere durch solche mit einer stärkeren 'Off-Target'-Kinase-Hemmung, von denen wir zuvor gezeigt haben, dass sie die Funktion der B-Zellen und die Antikörperantwort in vivo hemmen. "
Kurze Randbemerkung:
Natürlich würde man niemals nie nicht - jedenfalls nicht die Autoren - bereits eine Kontraindikation dabei vermuten, wenn man zeitgleich etwas unterdrückt und durch einen Schuss raufpushen will. (Einfach ein Pikser mehr! Wird schon gut gehen.)
Hier noch ein kurzer Offtopic-Knaller. der ebenfalls in dieser Studie erwähnt wurde:

“Die Analyse der neutralisierenden Antikörper wurde in allen Proben nach der Impfung unter Verwendung eines HIV-1-basierten Viruspartikels durchgeführt, das mit dem SARS-CoV-2 Wuhan Spike pseudotypisiert wurde, wobei alle Patienten positiv reagierten und eine mittlere ID50 von 445,5 (IQR 122-682) aufwiesen. Sieben von 16 (43,8 %) Patienten zeigten niedrige (50-200), 3/16 (18,8 %) mittlere (201-500) und 6/16 (37,5 %) hohe (501-2000) neutralisierende Titer3.”
Puh… Es gäbe wohl zu Metabolismus und den Mechanismen, die zum KRAS führen, noch sehr viel mehr zu sagen. Doch es ist unmöglich, dieses Thema in ein einziges Substack zu packen. Also musste ich mich dabei auf einen Themenkomplex fokussieren, wieso dieses Gen meiner persönlichen Interpretation nach, komplett aus den Fugen geschossen wird und gerade fleißig für Krebswachstum bei den Transfizierten sorgen dürfte.
Ich fasse bis hier hin nur nochmal zusammen, was eine der Hauptursachen sein wird:
Über B-Zellen und TLR (Deutsch)
Über B-Zellen und TLR (Englisch)

https://www.nature.com/articles/s41590-022-01168-4

Es wird die Kombination sein, welche sich aus einer zerstörten Zelloberfläche inklusive beispielsweise TLR, NLR (NOD-Like-receptors), Rig-Like-Receptors und SCAVENGER ergibt und den daraus entstehenden katastrophalen Folgekaskaden. Wir wissen mit Garantie, dass die LNP’s gerne durch die TLRs die Zelle transfizieren. Doch das bedeutet noch lange nicht, dass es nicht auch andere Rezeptoren in Mitleidenschaft ziehen wird. Und vergessen wir nicht, die direkten Membranschäden, wenn diese Fettkügelchen sich in die Zelle schweißen, als gäbe es kein Morgen.
https://journals.plos.org/plospathogens/article?id=10.1371/journal.ppat.1010830

Als man in dieser Studie beispielsweise das selbe Prinzip der LNPs für einen Influenzaimpfstoff und modifizierte RNA anwenden wollte, ging da auf dem TLR4 ganz schön die Post ab. Und die Autoren haben sich dabei nicht einmal den TLR7 und 8 auch nur ansatzweise angesehen.
Doch ein anderer wird natürlich auch die wtfRNA sein, die in die Zellen eindringt. Und auf eben diesen Mechanismus möchte ich mich in diesem Substack heute fokussieren. Ihr dürft also den Schaden, den diese wtfRNA anrichten wird zu den eh schon falsch getriggerten Kaskadenaddieren.
Und jetzt steige ich richtig in die Vollen ein:
Wer mit dem Begriff microRNA nicht wirklich etwas anzufangen weiß:
Über die dümmste Idee, die man haben kann (deutsch)
Über die dümmste Idee, die man haben kann (englisch)
Ich werde mich heute also eher auf die Schäden, direkt durch die wtfRNA ausgelöst, fokussieren.
Willkommen im echten Alptraum: Wenn all diese komplexen Kaskaden das Grundgerüst darstellen, aus dem Leben hervorgeht, sind die miRNAs quasi der Masterplan, durch welchen diese reguliert werden.
https://www.cell.com/molecular-therapy-family/nucleic-acids/pdfExtended/S2162-2531(18)30322-6

“Hier stellten wir fest, dass eine 100%ige Ψ-Substitution die Expression von IVT-mRNA im Vergleich zu unmodifizierter mRNA verdoppelte und das miRNA-vermittelte Silencing aller getesteten miRNA-Schalter leicht reduzierte, jedoch keine statistische Signifikanz erreichte. Ähnliche Ergebnisse wurden auch bei der m1Ψ-Modifikation beobachtet, von der bekannt ist, dass sie eine noch stärkere Proteinexpression als die Ψ-Modifikation hervorruft, und die das miRNA-abhängige Silencing der miR-126-3p-Schalter deutlich reduziert. Diese Daten stimmen mit früheren Berichten überein, die zeigen, dass der Einbau von Ψ oder m1Ψ in die IVT-mRNA die Translations Effizienz erhöht.”
(…)
“Ähnlich wie Ψ-modifizierte mRNA sind auch Ψ/m5C-modifizierte mRNAs dafür bekannt, dass sie die Proteinexpression verstärken und die Immunogenität verringern. Im Gegensatz zu Ψ-modifizierten miRNA-Schaltern bewirkte jedoch das Hinzufügen einer miRNA-Zielstelle zur 3'UTR ein miRNA-abhängiges Silencing von Ψ/m5C-modifizierten miR-126-3p-Schaltern ebenso effektiv wie bei Verwendung von vier Zielstellen. Außerdem wurde dieser Effekt nicht nur in HEK293-Zellen, sondern auch in HUVECs festgestellt. Der Anteil von m5C in den Zielstellen der getesteten miRNA-Schalter korrelierte nicht mit der miRNA-abhängigen Silencing-Effizienz, und die Silencing-Effizienz variierte auch in den verschiedenen Zelltypen, die in dieser Arbeit verwendet wurden. Die Nukleotidmodifikation verändert die Sekundär- oder Tertiärstruktur der mRNA, was die Zugänglichkeit der miRNA-Zielstellen auf eine switch-spezifische Weise verändern kann. Tatsächlich wurde berichtet, dass die Ψ-Modifikation die Proteinbindung an Konsensussequenzen in der mRNA verringert. Die Nukleotidmodifikationen könnten sich auch auf die Kinetik des Shuttles des miRNA-Schalters in oder aus den p-Körpern auswirken. Weitere Studien sind erforderlich, um die Wechselwirkung zwischen der miRNA-Zielsequenz und den durch Nukleotidmodifikationen verursachten Veränderungen beim miRNA-abhängigen Silencing zu untersuchen, insbesondere weil frühere Berichte eine Anreicherung von m5C am Ort der Argonaute-Bindung und Unterschiede in der Proteinexpression von modifizierter mRNA in verschiedenen Zelltypen gezeigt haben.”
Was die Autoren also beschrieben war eine Alterierung der Sekundär - und Tertiärstruktur der mRNA wo sie ihre m1Ψ-Modifikationen einfügten, welche damit auch die Zugänglichkeit für miRNAs ermöglichten.
Doch der wohl markanteste Satz war dieser:
"Diese Daten legen nahe, dass miRNA-Schalter, die auch nur eine vollständig komplementäre miRNA-Zielstelle enthalten, die Verfügbarkeit endogener kognitiver miRNA reduzieren und die Expression ihrer Zielgene beeinflussen."
Bedeutet nämlich, dass eben dann die Expression der Gene auch direkt affektiert ist. Sicherlich haben modRNA und Pfeyzer/ BioNTech dies berücksichtigt? Übrigens: Was bedeutet statistisch nicht signifikant bei in vitro Experimenten, wenn gut 70 Prozent der Studien, die beispielsweise in Nature erschienen sind, nicht reproduzierbar sind?
https://www.nature.com/articles/533452a

https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6599599/

“Aus früheren Berichten geht hervor, dass fast 90 % der Veröffentlichungen in der Krebsbiologie nicht reproduzierbar sind. Die niedrige Zahl wurde kürzlich durch fünf detaillierte Replikationsstudien in eLife bestätigt, die in Nature kommentiert wurden (Replikationsstudien bieten viel mehr als technische Details. Nature 2017;541:259-60). Während die Irreproduzierbarkeit oft menschlichen Faktoren zugeschrieben wird, die behebbar sind, könnte der Grund biologisch sein, und die Irreproduzierbarkeit ist für solche Studien immanent. Die geringe Reproduzierbarkeit, die die Vielfalt der evolutionären Wege der Tumorentstehung widerspiegelt, wird sich wahrscheinlich erheblich auf klinische Strategien auswirken.
Die Reproduzierbarkeit ist die Grundlage der experimentellen Wissenschaft. Zwar gibt es viele Faktoren, die verschiedene Forschungsbereiche in unterschiedlichem Ausmaß beeinträchtigen, doch scheinen krebsbiologische Studien eine Ausnahme zu bilden. In einem früheren Bericht konnten nur 6 von 53 veröffentlichten Ergebnissen in der Krebsbiologie bestätigt werden, was einer alarmierend niedrigen Reproduzierbarkeitsrate von 10 % entspricht. Dem Bericht zufolge ist eine solch niedrige Quote in der pharmazeutischen Industrie üblich.”
Ich habe Tränen gelacht, wer noch?!
Und jetzt mal überlegen:
https://academic.oup.com/nar/article/47/7/3353/5366472

“Insgesamt haben wir 2300 echte humane reife miRNAs extrapoliert, 1115 davon sind derzeit in miRBase V22 annotiert. Die experimentell validierten miRNAs werden dazu beitragen, hypothetische Targetome zu revidieren, indem falsch annotierte miRNAs verwendet werden.”
Da werden spezifische miR-switches bestimmt nicht weiter wild sein, wenn jede miR bestimmte gen- und RNA-regulatorische Funktionen übernimmt? Ach, Quatsch mit Sauce! Niemals nie nicht! Hoch wirksam, bestens erprobt und so sicher wie kein anderer Impfstoff weil wir direkt Milliarden Menschen damit beglückten. - Krebs sieht man schließlich so 2 - 3 Tage später, wenn er sich denn entwickeln würde. Doch genug von meinem Unverständnis, welches sich hier in Spott niederschlägt und zurück zur Quintessenz, wieso ich das KRAS so gruselig finde. Was die Autoren also feststellten, um noch einmal auf die Cell-Studie zu sprechen zu kommen, hing nicht nur von der Position der Substitutionen durch m1Ψ-Modifikationen ab, sondern war auch noch zellspezifisch zu beobachten.
https://tga.gov.au/sites/default/files/foi-2389-06.pdf

Was wissen wir über die Zielzellen des Transfektionsschusses? Ach ja… Lass dich überraschen, welche es denn sein werden. Phew. Nach dem ich nun also weiß, dass ich nicht einmal weiß, welche Zellen transfiziert werden und weiß, dass bei 2.300 miR ein vollmethylpseudouridylierter Viruscode nachhaltig reinkrachen wird…
Als nächstes überlegte ich nun also, ob es nicht doch vllt. ein paar Papers gäbe, die über miR-switches nach dem BNT162b2-Schuss berichtet hätten.
Steigen wir mit dem Schonkostpaper, welches dann endlich das Licht der Nature erblickte, ein:
https://www.nature.com/articles/s41541-022-00439-3

“Anschließend verglichen wir die miRNA-Spiegel mit spezifischen Antikörpertitern. Eine multiple Regressionsanalyse zeigt, dass die Spiegel von miR-132, miR-148a, miR-221 und miR-625-3p mit den Antikörpertitern zusammenhängen (Abb. 7a), während andere miRNA-Spiegel nicht mit den Antikörpertitern verbunden waren. Um die Korrelationen zwischen den einzelnen miRNAs und den Antikörpertitern weiter zu untersuchen, verglichen wir die miRNA-Spiegel zwischen den Gruppen mit hohen und niedrigen spezifischen Antikörpertitern. Interessanterweise stellten wir fest, dass die miR-148a-Spiegel in der Gruppe mit hohen Antikörpertitern signifikant niedriger waren (Abb. 7b). Als Nächstes führten wir eine multiple Regressionsanalyse durch, um die Zytokinspiegel mit den Serum-EV-miRNAs zu vergleichen. miR-126 und miR-451 waren mit den TNF-α-Spiegeln nach der ersten Dosis verbunden (Abb. 7c). Da TNF-α nach der ersten Dosis nicht induziert wurde, ist davon auszugehen, dass die TNF-α-Spiegel nach der ersten Dosis nur die basalen TNF-α-Spiegel widerspiegeln. Nach der zweiten Dosis wurden keine miRNAs mit den TNF-α- und IL-6-Spiegeln in Verbindung gebracht (Abb. 7d). Insgesamt deuten unsere Daten darauf hin, dass miR-92a-2-5p und miR-148a mit der Immunantwort nach der Impfung mit BNT162b2 in Verbindung gebracht werden können.”
“Interessanterweise ergab die multiple Regressionsanalyse, dass die miR-92a-2-5p-Spiegel in Serum-EVs einen Tag vor der ersten Dosis negativ mit den lokalen Scores nach der ersten und zweiten Dosis und auch mit den systemischen Scores nach der zweiten Dosis korreliert waren (Abb. 6b-e). Darüber hinaus korrelierte der miR-148a-Spiegel positiv mit den systemischen Ergebnissen nach der ersten Dosis (Abb. 6c). Im Gegensatz zur saisonalen Grippeimpfung standen die miR-451a-Spiegel in keinem Zusammenhang mit irgendwelchen Ergebnissen (Abb. 6b-e). Da die EV miR-92a-2-5p-Spiegel mit lokalen und systemischen Scores korreliert waren, untersuchten wir, welche Symptome mit miR-92a-2a-5p assoziiert waren. Unsere statistischen Analysen ergaben, dass die miR-92a-2-5p-Spiegel in den Serum-EVs bei Probanden mit Rötungen, Kopf- oder Gelenkschmerzen signifikant verringert waren (Abb. 6f). Systemische Scores von Frauen waren höher als die von Männern, aber es gab keinen Unterschied in miR-92a-2-5p Ebenen zwischen Frauen und Männern (ergänzende Abb. 5a, b), was darauf hindeutet, dass miR-92a-2-5p ist nicht mit geschlechtsspezifischen Unterschieden verbunden.”
“Interessanterweise waren miR-126, miR-132, miR-221 und miR-625-3p in unseren Regressionsanalysen auch mit den Antikörpertitern oder den Serum-Zytokinspiegeln nach der Impfung verbunden. miR-126 hemmt die VCAM-1-Expression in Endothelzellen und verringert dadurch die Adhärenz von Leukozyten an Endothelzellen, und es ist bekannt, dass miR-126 bei Sepsis erhöht ist.”
Na bitte. Klingt schon mal echt toll. Jaja, die Autoren meinten, es seien Biomarker. Doch die Switches passierten ja NACH der Pikse, richtig? Also gehen wir mal davon aus, dass eine Zelle nicht spontan sagt: “Puff!, ich mach jetzt einfach mal”, wenn es keinen Grund gäbe. Folglich wäre wohl der Schuss das Naheliegendste.
https://www.oncotarget.com/article/26263/text/

Was die Autoren dieser “Das sind doch nur Biomarker”- ”BNT162b2 macht ein bisschen Kopfschmerzen und Injektionsschmerzen”-Schwurbelei euch nicht verraten haben:
“Biochemische Stoffwechselwege wie der Glukose- und Aminosäurestoffwechsel waren in der metabolischen Tumoruntergruppe gestört, die auch einen hohen Anteil an KRAS- und APC-Mutationen aufwies und geringe Mengen an miR-143 und sechs Mitgliedern der let-7-Familie (let-7c, let-7e, miR-100, miR-125b-1, miR-125b-2 und miR-99a) exprimierte, die mit einer erhöhten KRAS-Expression einhergingen. Die metabolische Untergruppe war ebenfalls epithelialer Natur. Die Tumoren von Patienten mit dem schlechtesten Gesamt- und rezidivfreien Überleben gehörten zur mesenchymalen Untergruppe, die auch den höchsten Anteil an Tumoren im Stadium III und IV aufwies. MiRNAs, die sich zuvor als tumorsuppressiv erwiesen hatten (miR-148a, miR-192 und Mitglieder der miR-200-Familie), waren in mesenchymalen Tumoren herunterreguliert. Guinney et al. brachten die Herabregulierung von miR-192 und der miR-200-Familie mit dem Übergang von Epithel zu Mesenchym in Verbindung und die Herabregulierung von miR-148a mit der TGF-β-Signalübertragung und dem Matrixumbau, alles Prozesse, die in der Untergruppe der mesenchymalen Tumoren angereichert sind. Zu den TCGA-Proben mit eindeutigen molekularen Untergruppenzuordnungen, die von Guinney et al. definiert wurden, gehörten 40 (MSI-immun), 75 (kanonisch), 34 (metabolisch) und 66 (mesenchymal) Proben. Da unsere Daten zeigen, dass die mesenchymale Tumoruntergruppe mit tumorassoziierten Stromazellen angereichert ist, verwenden wir im Text den Begriff "stromale Untergruppe".”
Da sind wir auch schon wieder bei meinem KRAS. Ihr lest hoffentlich dasselbe wie ich und merkt, was für eine beschissene Idee es wäre, gleich mehrere von 2300 feinst ausbalancierten miRs umzuklappen, ohne überhaupt zu wissen, in welchen Zellen es passiert.
Und Let-7… Da kommen wir jetzt zu meiner personal favored Studie, dem Crimson-paper, welches ich nunmehr seit über einem Jahr studiere und mich immer noch “so klug wie hach” (Goethe - Faust) zuvor fühle:
https://crimsonpublishers.com/aics/fulltext/AICS.000552.php#/

“Nuclear Factor IB (NFIB) und v-myb avian Myelobastosis viral oncogene (MYB) wurden durch CovS-miR-48.1 mit miR-138- 5p bzw. CovS-miR-21 mit miR-155-5p gehemmt (Abbildung 4). Das MYB/NFIB-Fusionsproto-Onkogen erzeugt den MYB/NFIB-Transkriptionsfaktor und wurde bei einem hohen Prozentsatz der Patienten mit adenoidem zystischem Karzinom nachgewiesen. Außerdem wurde die Gruppe HMGA1 (High Mobility Group A1) durch CovS-miR-21 mit let- 7b-5p, let-7a-5p und miR-196a-5p unterdrückt (Abbildung 4). Da HMGA1 ein onkofötaler Transkriptionsfaktor ist und mit der Förderung und Metastasierung zahlreicher bösartiger Krebsarten in Verbindung gebracht werden kann, deuten beide Ergebnisse stark darauf hin, dass BNT162b2 nicht onkogen ist. Somit könnte die onkogene Aktivität von BNT162b2 durch von BNT162b2 abgeleitete miRNAs verhindert werden.”
Das Crimson Paper ist so irre. Schlussfolgerung: Wir haben krebsspezifische miRs durch andere krebspezifische miRs unterdrückt und feiern dies als vollsten, vollen Erfolg. Blöde nur, was unser Japaner dabei vielleicht vergessen hat zu überlegen:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4742387/

“Die Expression der let-7-Familie ist für das Entwicklungstiming und die Tumorsuppressorfunktion erforderlich, muss aber für die Selbsterneuerung von Stammzellen unterdrückt werden. Daher muss die let-7 miRNA-Biogenese sorgfältig kontrolliert werden. Um eine let-7 miRNA zu erzeugen, wird ein primäres Transkript durch die RNA-Polymerase II produziert und anschließend durch Drosha/DGCR8, TUTase und Dicer prozessiert. Da eine Dysregulation der let-7-Prozessierung schädlich ist, wird die Biogenese von let-7 durch zelluläre Faktoren wie die RNA-Bindeproteine LIN28A/B und DIS3L2 streng reguliert. In dieser Übersicht werden die biologischen Funktionen und die Biogenese von let-7 miRNAs erörtert, wobei der Schwerpunkt auf den molekularen Mechanismen der Regulierung der let-7-Biogenese bei Wirbeltieren wie der Maus und dem Menschen liegt.”
Könnte also vielleicht doch nicht der “sicher und wirksam”™ propagierte Effekt bei rauskommen. Um das mal anhand von Fallbeispielen zu demonstrieren, was eine solche kleine miR schon an den falschen Stellen angedockt anrichten kann, und um zeitgleich bei meinem KRAS-Thema zu bleiben:
https://www.nature.com/articles/tpj20109

Genetische Modulation der Let-7-MikroRNA, die an die 3′-untranslatierte Region von KRAS bindet, und Überleben von Patienten mit metastasierendem Kolorektalkarzinom, die mit Cetuximab-Irinotecan als Heilmittel behandelt werden

Ein Let-7 MicroRNA SNP im KRAS 3′UTR ist prognostisch bei Darmkrebs im Frühstadium
Wieso ist der Mechanismus dahinter so wichtig? Wir müssen bestmöglich verstehen, was wodurch ausgelöst wurde, um bestmöglich noch lindern zu können.
Darmkrebs also… Und das war nur die Let-7 microRNA.
https://www.nature.com/articles/s41419-017-0243-9

Kommen wir nun also langsam dazu, wieso es so wichtig war, faktisch die miR-switches und die Signalkaskade beim KRAS zu beleuchten:
“Das onkogene KRAS induziert die Entstehung und Entwicklung von Tumoren, indem es die Genexpression über verschiedene molekulare Mechanismen moduliert. MicroRNAs (miRNAs) sind kleine nicht-kodierende RNAs, die als Hauptakteure in der Tumorentstehung etabliert sind.”
Hierbei gilt es zu berücksichtigen, dass die Autoren nur ein paar spezifische Beispiele aufführten. Doch warum der Fokus auf dem KRAS und nicht etwa auf dem Gen, zu dem es führen könnte, beispielsweise dem p53?:
Weil sich hinter dem KRAS und der Signalkaskade soviel mehr verbirgt.
Erinnert ihr euch an mein Substack über die CD4+-T-Helferzelllinie und die Th-17?:

https://www.nature.com/articles/s41467-020-19288-6

Schaut mal, was das KRAS alles triggert. Und hier haben wir sie also: Die Kaskaden zu einer dysregulierten Th-17, ein dysreguliertes NF-kB, welches über Monate TNF-alpha zurückfeuern wird und viele weitere nette Sachen. Unter anderem auch ICAM und das Interleukin 8. Direkt und ohne Umschweife
Ja, aber, aber das könnten doch zig andere Mechanismen sein. Wieso bin ich denn bloß so versteift auf das KRAS?:
Vergleichen wir doch mal, was wir definitiv wissen und was nicht, weil keine dementsprechenden Untersuchungen gemacht werden:
https://www.cell.com/trends/biochemical-sciences/fulltext/S0968-0004%2813%2900203-X

https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8163516/

https://jmedicalcasereports.biomedcentral.com/articles/10.1186/s13256-022-03607-0

https://www.mdpi.com/2227-9067/9/1/29

https://link.springer.com/article/10.1007/s10067-022-06097-z


Gut, gut, den Pankreas haben wir uns nun also schonmal angesehen:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8976114/

Praktischer Weise konnten die Autoren natürlich schlussfolgern:
”Darüber hinaus wurden Symptome von Menstruationsstörungen nach der Impfung nicht mit PCOS, Schilddrüsenstörungen, Uterusmyomen, Endometriose und Adenomyose in Verbindung gebracht (p > 0,05).”
Nun ja, liebe Berufssöldner, die ihr euch da erbarmt habt, überhaupt über die Menstruationsunregelmäßigkeiten zu schreiben:
Wir haben also den Link zur Th17 (siehe mein oben erwähntes Substack zur CD4+) und diese wiederum direkt reguliert durch das KRAS….
Gut dass die Methodiken der aktuellen “die Wissenschaft”™ auf Assoziationen und nicht etwa akribischen und methodisch sauberen Untersuchungen beruhen. Und da man praktischerweise auch beides kaufen kann, in dem man beispielsweise selektive Methodiken wählt, nur die Wunschresultate publiziert, etc, etc, muss man sich auch nicht weiter Sorgen um die Schlussfolgerung: “Sicher und wirksam”™ machen.
Dabei hätte ich schwören können, dass auch Menstuationsblutungen irgendwie was mit dem KRAS zu tun gehabt hätten?
War da nicht ne Kleinigkeit mit SIRT1 und BCL6 (ENZYME)?:
https://pubmed.ncbi.nlm.nih.gov/28754906/

“Endometriose ist eine entzündliche Erkrankung, die mit Progesteronresistenz und Zellproliferation einhergeht und zu Schmerzen, Unfruchtbarkeit und Schwangerschaftsverlust führt. Wir haben zuvor die Phosphorylierung von STAT3 im eutopen Endometrium von unfruchtbaren Frauen mit dieser Erkrankung nachgewiesen, was zu einer Überexpression des Onkogens BCL6 und einer Stabilisierung des Hypoxie-induzierten Faktors 1 alpha (HIF-1α) führt. Hier berichten wir über eine koordinierte Aktivierung von KRAS und eine Überexpression von Sirtuin 1 (SIRT1), einer Histondeacetylase und einem Gen-Silencer, im eutopen Endometrium von Frauen mit Endometriose während des gesamten Menstruationszyklus. Bei Mäusen mit konditionaler Aktivierung von KRAS in den PGR-positiven Zellen ist die SIRT1-Expression im Endometrium im Vergleich zu Kontrollmäusen erhöht. Die Expression von Progesteronrezeptor-Zielgenen, einschließlich der Gene des indischen Hedgehog-Signalwegs, ist bei den mutierten Mäusen deutlich herabreguliert. SIRT1 wird mit BCL6 in den Zellkernen der betroffenen Individuen kolokalisiert, und beide Proteine binden an den Promotor von GLI1, einem kritischen Vermittler der Progesteronwirkung im indischen Hedgehog-Signalweg, und unterdrücken diesen, wie eine ChIP-Analyse ergab. Im eutopen Endometrium ist die GLI1-Expression bei Frauen mit Endometriose reduziert. Zusammengenommen deuten diese Daten darauf hin, dass KRAS, SIRT1 und BCL6 im eutopen Endometrium von Frauen mit Endometriose koordiniert überexprimiert sind und wahrscheinlich an der Pathogenese der Endometriose beteiligt sind.”
https://academic.oup.com/humupd/article/27/6/1086/6299969
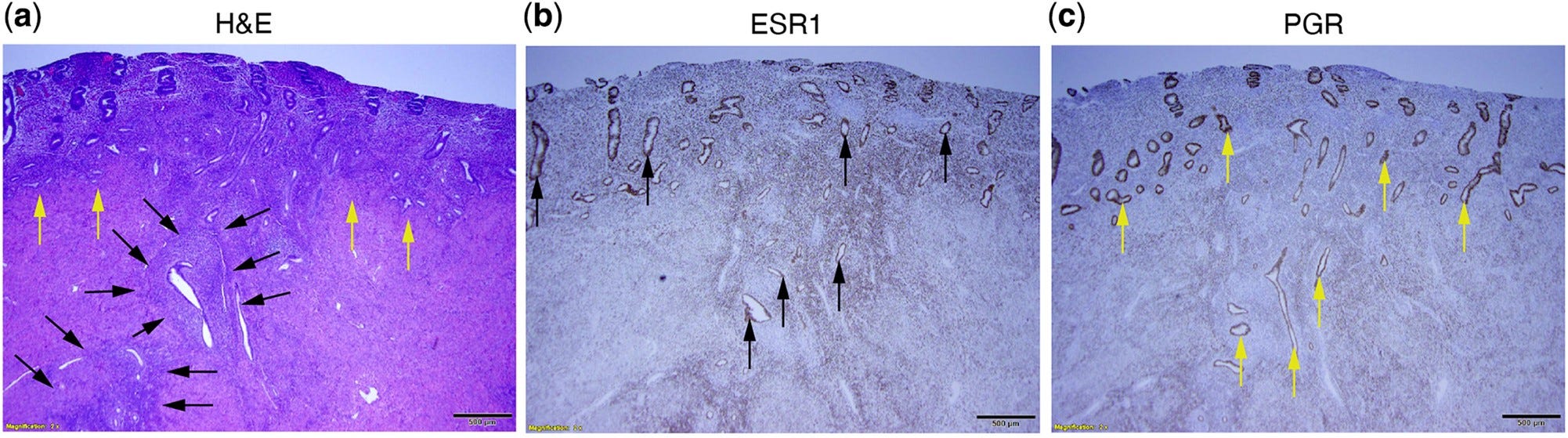
"Dazu gehören Wege, die eine erhöhte Östrogenbiosynthese, einen verringerten Östradiol-Stoffwechsel, einen einzigartigen, von Östrogenrezeptor beta (ESR2) gesteuerten Entzündungsprozess und eine Progesteronresistenz aufgrund einer verringerten Progesteronrezeptorexpression begünstigen. Da die Adenomyose einen einzigartigen östrogengesteuerten Entzündungsprozess und eine Progesteronresistenz aufweist, erörtern wir die Wechselwirkungen zwischen diesen molekularen Merkmalen und den Signalwegen, die durch die neu entdeckten KRAS-Mutationen ausgelöst werden.
(...)
Es gibt Hinweise darauf, dass KRAS-Mutationen zum Teil für zuvor beobachtete Phänomene wie das verlängerte Zellüberleben und die Progesteronresistenz bei Adenomyose verantwortlich sein könnten."
Hm. Hat bestimmt nichts weiter mit diesem “Phänomen”™ zu tun. Alles so mysteriöse Zufälle, welche aus purstem purem Zufall genau in die Charakteristiken fallen, was KRAS so alles tolles mit Zellen anstellt.
Ich acker mich mal durch noch zwei weitere Beispiele, um es zu verdeutlichen.
Ihr erinnert euch an weiter Oben, wo ich auch das TNF-alpha anschnitt?:
https://www.science.org/doi/10.1126/sciimmunol.abl5344

Wie sollte das möglich sein, ohne dass die Zellen frühzeitig in Apoptose untergehen? Denn TNF-alpha muss man sich als ein wirklich extrem neurotoxisches Zytokin vorstellen, welches eigentlich nur ein kurzes Signal an die T-Zellen liefern soll. Zu lange und zu hoch dosiert, so wie nach dieser Transfektion… oha…
https://www.nature.com/articles/cdd201019

“Dieser TNF-α-induzierte Zelltodweg wird normalerweise durch die gleichzeitige Aktivierung von NF-κB blockiert.”
Immer daran denken: Sicher und wirksam.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9319879/
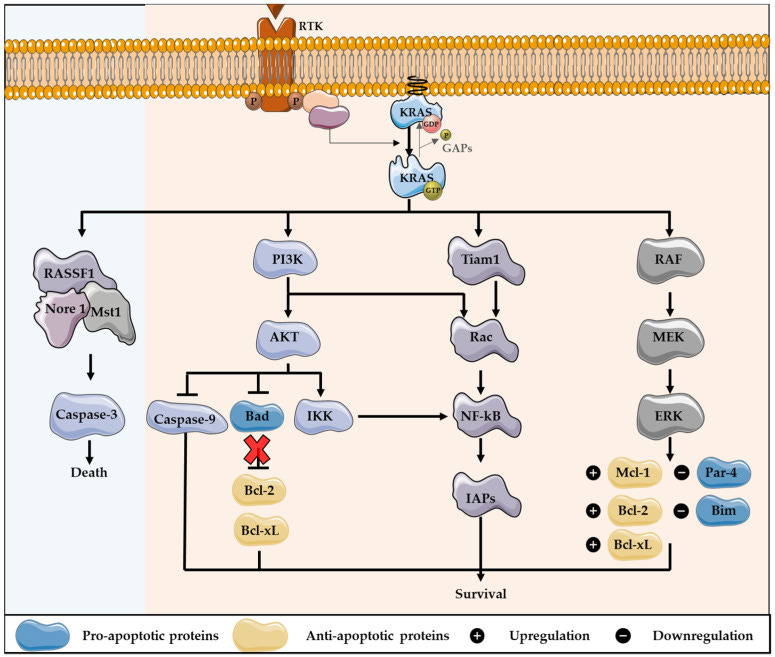
“KRAS, ein Mitglied der RAS-Proteinfamilie, spielt durch die Regulierung mehrerer nachgeschalteter Effektoren eine wichtige Rolle bei Autophagie und Apoptose. In Krebszellen führen KRAS-Mutationen zu einer konstitutiven Aktivierung dieses Onkogens, das die Zellproliferation anregt, die Autophagie induziert, die Apoptose unterdrückt, den Zellstoffwechsel verändert, die Zellmotilität und -invasion verändert und die Mikroumgebung des Tumors moduliert.
(…)
Ein weiterer wichtiger KRAS-Effektor ist der RAL-Guanin-Nukleotid-Dissoziationsstimulator (RALGDS), der RAS-ähnliche (RAL-)GTPasen aktiviert und überlebensfördernde Funktionen hat sowie die Zellzyklusprogression fördert. Darüber hinaus interagiert KRAS mit dem T-Lymphom-Invasions- und Metastasenprotein 1 (Tiam1), einer GTPase der Rho-Familie, die an der Entwicklung von RAS-gesteuerten Tumoren, der Wachstumstransformation, der Förderung des Zellüberlebens, der Aktivierung der mitogen-aktivierten Proteinkinase c-Jun amino-terminal kinase (JNK) und der Aktivierung des Transkriptionsfaktors NF-kB beteiligt ist. RAS bindet auch an PLCε 44, eine Phospholipase-C-Isoform, die für die RAS-vermittelte Produktion des Membranlipids Diacylglycerin (DAG) verantwortlich ist, was zur Freisetzung von Kalzium und zur Aktivierung der PKC-Signalkaskade führt, die am Überleben, der Proliferation und der Kalziummobilisierung beteiligt ist.”
Hier gibt es nichts zu sehen. Bitte gehen Sie ganz schnell weiter.
https://www.sciencedirect.com/science/article/pii/S002192581749510X

Den hier halte ich kurz und frage nur mal mit einem verstörten Grinser auf meinen Lippen: Könnte KRAS auch der Schlüssel zum p53 sein?
Man weiß es ja immer nicht, man munkelt ja nur…
Nachtrag 17.10.2022:
Vielen dank, lieber DoorlessCarp für diese fiese Studie:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5630394/

“Der prognostische Wert von RAS-Mutationen wurde bei akuter myeloischer Leukämie (AML) systematisch untersucht. Die klinische Bedeutung der RAS-Expressionen bei AML ist jedoch nur unzureichend bekannt. Um die klinische Bedeutung zu untersuchen, analysierten wir die KRAS- und NRAS-Expressionen bei 143 de novo AML-Patienten mittels quantitativer Echtzeit-PCR. Die KRAS- und NRAS-Expressionen waren bei AML-Patienten signifikant hochreguliert. KRAS- und NRAS-Mutationen wurden bei 4% (6/143) bzw. 8% (12/143) dieser Patienten festgestellt. Es wurde jedoch kein signifikanter Zusammenhang zwischen RAS-Mutationen und -Expressionen festgestellt. Eine hohe KRAS-Expression war mit höherem Alter, höheren weißen Blutkörperchen und tendenziell höheren Blutplättchen verbunden, während eine hohe NRAS-Expression nur mit höherem Alter korreliert war. Die Komplettremissionsrate und das Gesamtüberleben von AML-Patienten wurden durch die KRAS-Überexpression beeinträchtigt, nicht jedoch durch die NRAS-Überexpression. Die multivariate Analyse ergab, dass KRAS bei zytogenetisch normaler AML (CN-AML) als unabhängiger prognostischer Prädiktor fungierte. Darüber hinaus wurde der prognostische Wert der KRAS-Expression anhand der veröffentlichten Daten aus Gene Expression Omnibus-Datensätzen validiert. Bei den Follow-up-Patienten nahm die KRAS-Expression im Vergleich zur NRAS-Expression in der CR-Zeit tendenziell ab, und sowohl die KRAS- als auch die NRAS-Expression waren in der Rezidivzeit signifikant erhöht. Unsere Ergebnisse zeigen, dass RAS-Überexpression und -Mutationen bei AML häufig vorkommen und ein potenzielles therapeutisches Ziel darstellen. Eine von RAS-Mutationen unabhängige KRAS-Überexpression führte zu einer ungünstigen Prognose bei CN-AML.”
Was diese Studie zeigte, ist, dass auch unabhängig von RAS-Mutationen alleine die Überexpression von KRAS ausreichte, um bei Leukämie die Prognosen deutlich zu verschlechtern. Diese Studie war es mir doch wert, noch angeschnitten zu werden, da sich damit die Frage auftut, ob es nur - so wie in den anderen Studien gezeigt - bei Mutationen zu katastrophalen Konsequenzen führen würde? (Die Mutationen haben natürlich auch mit der gesteigerten Expression gute Chancen, zu passieren.) Das werden noch spannende Tage.
Nachtrag ende
Mir dampft der Kopf. Euch auch?
Ich denke, ich werde so langsam zum Ende kommen. Wie bereits oben erwähnt: Das KRAS ist eines der komplexesten Elemente bei den Kaskaden und hat so unvorstellbar viele Verstrickungen. Ihr habt gesehen, wohin es führt?: RAF, MEK, ERK…. Und ja. Jedes einzelne davon wäre noch einmal so ein Höllentrip.
Ich betone noch einmal, dass dies eine hypothetische Überlegung, angepasst an die aktuell real beobachtbaren Daten der Geburtenrückgänge, Nebenwirkungen und vermutlich noch kommenden Nebenwirkungen, ist, die nicht zwingend eintreffen wird, aber nach Ockhams Rasiermesser eine der logischsten Erklärungen für viele dieser Prozesse wäre.
Ich schließe dieses Mal jedoch etwas anders und nicht ganz so zynisch. Für diese Studie bedanke ich mich aus tiefstem Herzen bei unserem DoorlessCarp (auch hier auf Substack zu finden):
Quizfrage: Könnte man die KRAS-Regulation eventuell via BAICALEIN ein wenig verbessern? Fragen Sie doch einfach mal Ihren freundlichen Onkologen aus der Geldmachabteilung:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5929448/

In diesem Sinne: Narfige Grüße, euer Genervter Bürger.
Meine Güte ...was eine geniale Arbeit. Vieles muss ich als “alte Biologin” selber noch lernen müssen. Und die Arbeit mehrmals studieren. Danke - unvaxxed...
holy moly narf mich noch durch den ersten Paragraph.
"GDP-gebunden, was inaktiv ist, und GTP-gebunden, was aktiv ist und Signale durch Interaktion mit verschiedenen nachgeschalteten Effektoren überträgt. "
sollte hier hinter dem zweiten GTP *un*-gebunden stehen?
liebe Grüße und weiter zur Lektüre