

Ein Fass ohne Boden - über Toll-Like-Rezeptoren und B-Lymphozyte
Antikörper! Ganz, ganz, ganz, ganz viele bitte!

https://www.nature.com/articles/s41579-021-00573-0

Dann stelle ich mich heute mal dem Thema Antikörper. Ein Höllenthema und dies wird ein sehr langer und sehr umfangreicher Artikel. Ich werde zeigen, dass Antikörpertiter bei einigen Viren ein echter Nobrainer sind und wie kritisch die Rolle der B-Zellen in viel mehr Prozessen, als der Produktion von in diesem Fall nutzloser Antikörper gegen ein einziges Protein (das Spike), sind (von dem gerade mal nur die Oberfläche gebunden wird). Noch dazu, wenn es sich um funktionsfähige Sequenzen des Glykoprotein 120 und Glykoprotein 41 im Epitop handelt.

Dieser Artikel wird vermutlich sehr lang und sehr intensiv. Und es ist unmöglich, mir zu folgen, ohne meinen Artikel über die CD8+/CD4+/B-Zellen allgemein und über die CD4+ im Speziellen gelesen zu haben, da ich hier zwingend auch kurz auf die Tfh eingehen muss. Und wenn man mit dieser Abkürzung nichts anzufangen weiß, darf man sich sicher sein - so laienfreundlich ich es auch wieder versuche, zusammenzufassen - dass man nicht mehr folgen kann.
Hier also nochmal die wichtigsten Substacks in Reihenfolge, um mit mir weitere Überlegungen anstellen zu können:
Deutsch:
Über T- und B-Zellen und negative Effektivität
https://genervter.substack.com/p/uber-negative-effektivitat-und-zerschossene (English

Über Signalwege und Interleukine

CD8+
https://genervter.substack.com/p/in-der-hohle-des-lowen?utm_source=%2Fsaved&utm_medium=reader2

CD4+
https://genervter.substack.com/p/am-abgrund?utm_source=/saved&utm_medium=reader2

Steigen wir also voll ein
Noch mal zur Erinnerung: Die Stelle auf der Oberfläche eines Antigens, die von einem Antikörper erkannt wird, wird als Epitop bezeichnet. An dieses Epitop bindet ein Antikörper mit dem sogenannten Paratop, der gegenüberliegenden passgenauen Stelle auf der Oberfläche des Antikörpers.
Schema der Entstehung:
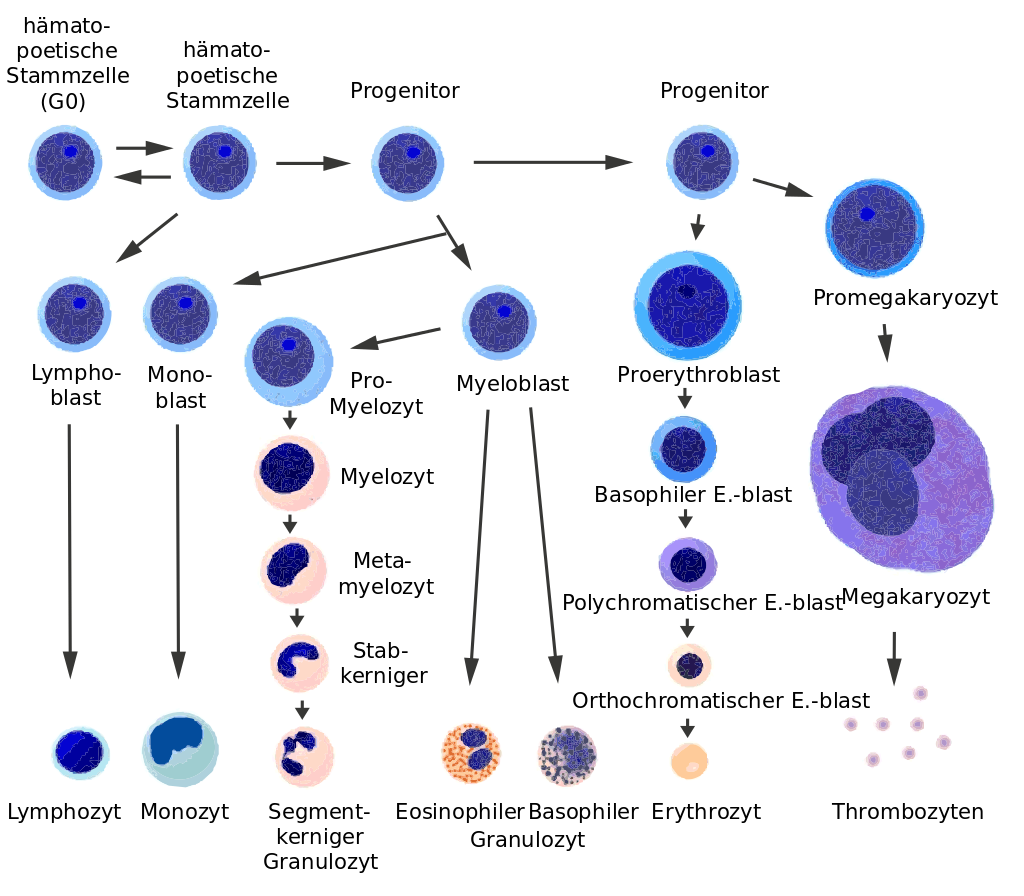
B-Zellen (B lymphozytische Zellen) , welche die antigenspezifischen Rezeptoren auf ihrer Oberfläche präsentieren, werden - genau, wie die T-Zellen - aus neutralen hämatopoetischen Stammzellen gebildet und differenziert. (Bild Wikipedia)
https://www.wikiwand.com/en/Haematopoiesis
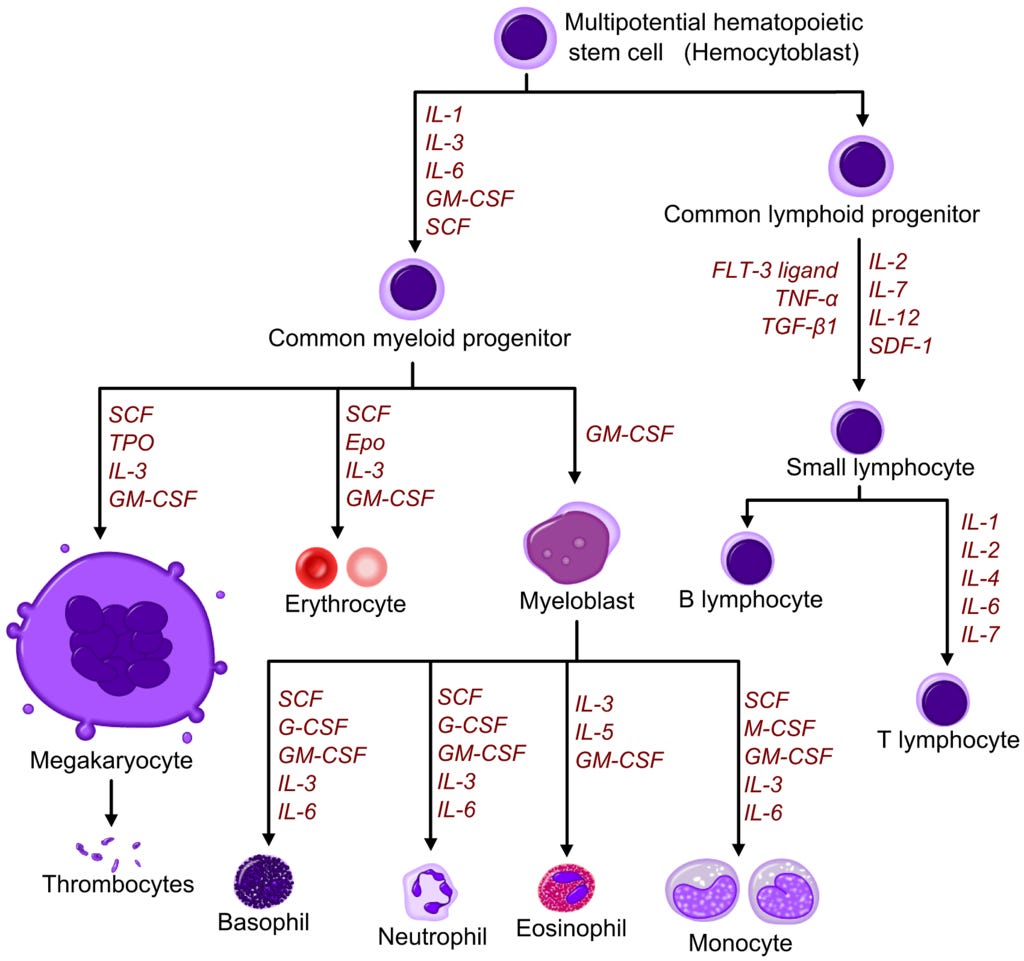
Wer die Differenzierung noch besser verstehen will:
Hier ist ein wirklich schönes Lernvideo.
https://www.nature.com/articles/nri3857
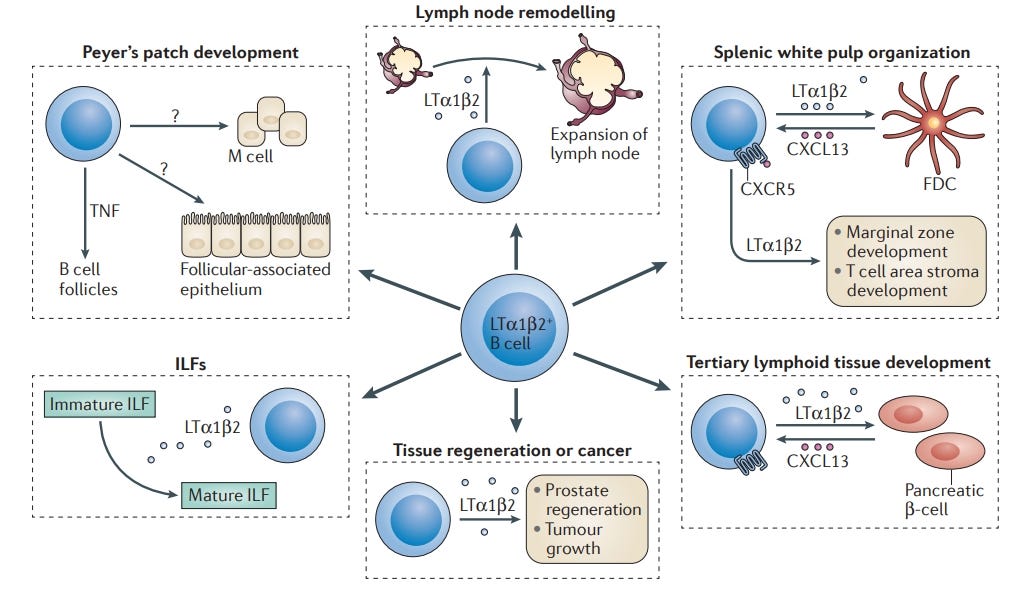
“Die Zytokinproduktion durch B-Zellen ist für zahlreiche Aspekte der Immunität von Bedeutung. Aus B-Zellen stammende Zytokine, einschließlich Lymphotoxin, sind für die Ontogenese, Homöostase und Aktivierung sekundärer lymphatischer Organe sowie für die Entwicklung tertiärer lymphatischer Gewebe an ektopischen Stellen von wesentlicher Bedeutung. Andere von B-Zellen stammende Zytokine wie Interleukin-6 (IL-6), Interferon-γ und Tumornekrosefaktor beeinflussen die Entwicklung von Effektor- und Gedächtnis-CD4+-T-Zellantworten. Schließlich können B-Zellen entzündliche Immunreaktionen regulieren, vor allem durch die Bereitstellung von IL-10 und IL-35. Diese Übersicht fasst diese verschiedenen Rollen der Zytokin-produzierenden B-Zellen in der Immunität zusammen und erörtert die Gründe für eine gezielte Behandlung dieser Zellen in der Klinik”

Was mir dabei wieder bewusst wird: Ich komme nicht umher, mich früher oder später mit dem Alptraum falsch getriggerter Zytokine noch wesentlich intensiver zu beschäftigen. Doch heute noch nicht. Dazu brauche ich wohl noch sehr viel Zeit, da dies ohne ein Mindestverständnis für organische Chemie und Biochemie nicht zu bewerkstelligen sein wird. Wer hätte nur damit gerechnet, dass B-Zellen nicht nur zum Antigenbinden durch ihre Oberflächerezeptoren dienen? (Jeder, der auch nur einen Moment nachdenkt, eventuell?)

Und hier kommt auch schon der erste Witz für B-Zellen:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7094289/
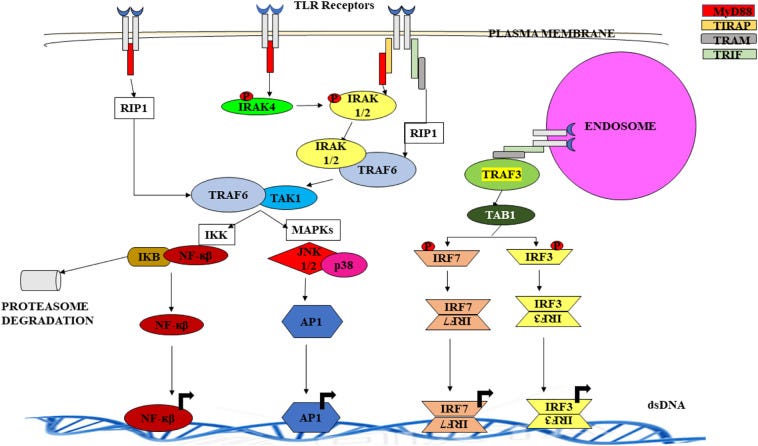
“TLR7 wird mit der Produktion von IFN Typ-1 in Verbindung gebracht, das für die Beseitigung von Viren wie dem Coronavirus des schweren akuten Atemwegssyndroms (SARS CoV) wichtig ist. TLR4 und MyD88 spielen ebenfalls eine schützende Rolle bei einer SARS-Infektion. TLR7 spielt auch bei einer Influenza-Infektion eine wichtige Rolle. Sie werden auf pDCs exprimiert und vermitteln über die MyD88-abhängige IFN-Induktion eine zellspezifische Immunantwort. Ein Isotypwechsel der B-Zellen wird ebenfalls bei einer Influenza-Infektion beobachtet und durch MyD88 und IPS-1 (Adaptormolekül, das am zytosolischen RIG-I-Weg beteiligt ist) vermittelt. Es wurde festgestellt, dass TLR3 in menschlichen alveolären Bronchial- und Epithelzellen während einer Influenza-Infektion stark hochreguliert wird. Darüber hinaus wurde festgestellt, dass auch RIG-I, NLRP3 und NOD-2 mit der Immunantwort gegen Influenza-Infektionen in Verbindung stehen.”
Puh! Eigentlich wollte ich doch nur über B-Zellen reden? Na gut. Dann stellen wir uns heute doch mal noch in dem Kontext dem Problem der Toll-Like-Rezeptoren (TLR):
Dazu muss ich zunächst den Begriff Signalkaskade verstehen:
”Unter einer Signalkaskade versteht man in der Molekularbiologie einen mehrstufigen Prozess an dem verschiedene Moleküle (Enzyme und/oder Signalmoleküle) beteiligt sind. Dabei wird das Signal ggf. in seinem Übertragungsverlauf verstärkt. Der Begriff wird auch in der Medizin verwendet, z.B. in der Onkologie beim Bezug auf onkogene Signalwege.”

”Toll-like Rezeptoren, kurz TLRs, sind eine Klasse von Pattern-Recognition-Rezeptoren (PRR). Sie erkennen Pathogene über PAMPs und spielen damit bei der angeborenen Immunität eine zentrale Rolle.
Pattern-Recognition-Rezeptoren funktionieren wie die klassischen Rezeptoren der gesamten Immunantwort: Sie können beispielsweise als Rezeptoren bei der Phagozytose dienen, um Erreger erstmals zu erkennen. Daraufhin können die Antigene auf den Oberflächen dieser phagozytierenden Zellen dargestellt werden und so weitere Signalkaskaden der Immunantwort auslösen.
PRRs erkennen außerdem auch sogenannte damage-associated molecular patterns (DAMPs). Dabei handelt es sich um körpereigene, normalerweise intrazelluläre Moleküle, die bei Schädigung einer Zelle ins Extrazelluläre gelangen. Durch die Bindung an die Rezeptoren wird eine Entzündungsreaktion ausgelöst. DAMPs sind z.B. Hitzeschockproteine, ATP oder das Protein HMGB1 ("High-Mobility Group Protein B1").”
Uff. Jetzt folgt erstmal ein langer aber wirklich guter Crahskurs in Sachen Toll-Like-Rezeptoren. Das wird noch wichtig, da ich untersuchen will, wie die B-Zellen nach dem Transfektionsschuss reagieren und was für Antikörper geformt werden.
https://www.mdpi.com/2218-273X/11/9/1291/htm


“Was bewirken Toll-Like-Rezeptoren?
Eine kleine Anzahl von Toll-ähnlichen Rezeptoren kann ein breites Spektrum menschlicher Krankheitserreger sowie eine Vielzahl anderer Moleküle, die auf Gewebeschäden hinweisen, durch einen Prozess namens Mustererkennung erkennen. Diese Rezeptoren leiten zwei Teile der Immunreaktion ein - die angeborene und die adaptive Reaktion -, die zusammenarbeiten, um Infektionen bei Säugetieren zu bekämpfen. Die angeborene Reaktion bietet sofortigen Schutz. Sie ist jedoch relativ unspezifisch in ihrer Art, Krankheitserreger anzugreifen, was zu einer Schädigung des gesunden Gewebes führt, wenn die angeborene Immunantwort zu lange anhält. Die adaptive Reaktion hingegen erzeugt Antikörper-sezernierende B-Zellen und zytotoxische T-Zellen, die spezifisch und effizient gegen Krankheitserreger vorgehen. Leider dauert die Entwicklung dieses Prozesses länger als bei der angeborenen Reaktion.
Da die Toll-like-Rezeptoren als erste Antwort auf Gefahrensignale fungieren, sind sie von zentraler Bedeutung für die Forschungsbemühungen zur Bekämpfung von Infektions- und Entzündungskrankheiten. Neue Strategien zur Beeinflussung von Immunantworten hängen vom Verständnis der Zellbiologie der Toll-like-Rezeptoren ab, einschließlich ihrer Struktur, Zelllokalisierung, Signaltransduktionswege und Expressionsmuster.
Mustererkennung und Maut
Menschliche Zellen haben nur etwa 25.000 proteinkodierende Gene. Es ist also unmöglich, für jede Art von Virus, Bakterium, Protist oder Pilz ein anderes Gen (und einen anderen Rezeptor) zu haben. Wie kann der Körper dann alle Arten von Krankheitserregern erkennen, die eine Gefahr darstellen, auch solche, denen er noch nie begegnet ist? Charles Janeway schlug 1989 vor, dass Zellen die Mustererkennung nutzen, um Krankheitserreger zu erkennen (Janeway 1989). Mit anderen Worten: Rezeptoren binden an strukturelle Formen oder Muster, die als PAMPs (pathogen-associated molecular patterns) bezeichnet werden und die in ganzen Gruppen von Krankheitserregern, nicht aber im Wirt vorhanden sind. Nach Janeways Theorie können die Rezeptoren eine bestimmte Mikrobe nicht genau identifizieren, aber sie können sie als einen fremden Organismus erkennen.
Die ersten menschlichen Mustererkennungsrezeptoren wurden zehn Jahre nach Janeways Vorschlag identifiziert. Der Durchbruch wurde durch eine frühere Entdeckung bei der Fruchtfliege Drosophila ermöglicht. Jahrzehntelang hatten Forscher Drosophila zur Identifizierung von Entwicklungsmutationen verwendet, aber Drosophila wurde nicht als geeignetes Modell für die menschliche Immunität angesehen, da es bei Insekten keine adaptive Reaktion gibt (Beck & Habicht 1996). Deutsche Wissenschaftler identifizierten ursprünglich das Toll-Gen als den Ort der Mutationen, die zu bizarr aussehenden Fliegen führten. (Sie riefen aus, dass ihre Ergebnisse "Toll!" seien, was auf Englisch "Great!" bedeutet.) Die Klonierung des Gens zeigte, dass es für einen Membranrezeptor kodiert (Hashimoto, Hudson & Anderson 1988).
In einer Studie aus dem Jahr 1996 wurde berichtet, dass Toll-Mutationen mit Funktionsverlust Drosophila sehr anfällig für Pilzinfektionen machen und dass Mutationen mit Funktionsgewinn zu einer erhöhten Produktion bestimmter antifungaler Proteine führen (Lemaitre et al. 1996). Vergleiche von Toll-Mutationen mit Mutationen in anderen Genen führten zu der Schlussfolgerung, dass der Toll-Rezeptor eine dominante Rolle bei der Erkennung von Pilzinfektionen und der Auslösung der angeborenen Immunantwort spielt. Diese aufregende Entdeckung lieferte den Forschern den Anhaltspunkt, den sie brauchten, um menschliche Pathogenrezeptoren zu finden. Anhand der Aminosäuresequenz von Toll suchten sie in der Datenbank des Humangenomprojekts nach verwandten Sequenzen und identifizierten Toll-ähnliche Rezeptoren (Medzhitov, Preston-Hurlburt & Janeway 1997; Rock et al. 1998).
Verständnis der Toll-Like-Rezeptoren
Schematische Darstellungen zeigen die Moleküle in sechs mikrobiellen Krankheitserregern, die von spezifischen Toll-like-Rezeptoren gebunden werden. Die sechs dargestellten Krankheitserreger sind: RNA-Viren, DNA-Viren, Gram-positive Bakterien, Gram-negative Bakterien, Pilze und Protisten.
Toll-like-Rezeptoren (TLRs) erkennen Mikroben durch Bindung an pathogen-assoziierte molekulare Muster. Abkürzungen: Lipopolysaccharid (LPS), Lipoteichosäure (LTA), Lipoproteine (LP), Glycophosphatidylinositol (GPI). Andere Mustererkennungsrezeptoren für Krankheitserreger wurden identifiziert, wie z. B.: Transmembranrezeptoren für C-Typ-Lektine (CLRs), die Pilze erkennen; sekretierte Rezeptoren (Collectins, Ficolins und Pentaxins), die die angeborene Abwehr mit Komplement und Phagozytose aktivieren; zytosolische RIG-1-ähnliche Rezeptoren (RLRs), die Viren erkennen; und zytosolische Rezeptoren mit Nukleotidbindungsdomäne und leucinreichen Wiederholungen (NLRs), die Krankheitserreger und Stresssignale erkennen.
Wissenschaftler stellten die Theorie auf, dass Toll-ähnliche Rezeptoren (TLRs) aufgrund ihrer Ähnlichkeit mit der Aminosäuresequenz von Toll eine Immunreaktion auf Krankheitserreger auslösen würden. Aber wie konnte diese Theorie experimentell bestätigt werden? Mausmodelle lieferten die notwendige Bestätigung, indem sie es ermöglichten, die Funktion der TLRs in vivo (in einem ganzen Organismus) zu untersuchen.
Mausmodelle werden zur Untersuchung einer Reihe von menschlichen Entzündungskrankheiten verwendet, die durch eine überaktive angeborene Immunantwort verursacht werden, die zu gefährlichen Entzündungen und Gewebezerstörungen führt. So ist beispielsweise die Sepsis eine schwere Krankheit, bei der der Blutkreislauf mit Bakterien überschwemmt wird, was zu Entzündungen im gesamten Körper führt. Jedes Jahr sterben in den Vereinigten Staaten mehr als 200.000 Menschen daran. Bei der Entwicklung von Mausmodellen für Sepsis werden Mäusen Bakterien injiziert, um eine Entzündung auszulösen. Der Schweregrad der Krankheit wird dann mit kontrollierten und standardisierten Tests gemessen. Wissenschaftler wissen seit vielen Jahren, dass isolierte bakterielle Komponenten wie Lipopolysaccharid (LPS) ganze Bakterien ersetzen können, um eine Sepsis auszulösen. LPS ist in den Zellwänden aller gramnegativen Bakterien vorhanden, die eine große Gruppe von Krankheitserregern darstellen. Es ist daher ein Beispiel für die PAMPs von Janeway. Darüber hinaus wiesen Poltorak et al. (1998) nach, dass Mäuse mit einer defekten Version von TLR4 völlig resistent gegen LPS-induzierte Sepsis sind. Damit wurde nachgewiesen, dass TLR4 der Rezeptor für LPS ist und dass er eine angeborene Immunreaktion nach LPS-Stimulation auslöst.
Wissenschaftler wissen heute, dass der Mensch über mindestens zehn verschiedene TLRs verfügt, die zusammen ein breites Spektrum von Krankheitserregern erkennen (Abbildung 1). Die TLRs 1, 2, 4, 5 und 6 binden an Komponenten der mikrobiellen Zellwände und Membranen, die nur bei Krankheitserregern vorkommen. Die am besten charakterisierten Liganden sind bakteriell, darunter LPS und Lipoteichonsäure aus Zellwänden, Lipoproteine aus der Zellmembran und ein struktureller Bestandteil bakterieller Geißeln, das Flagellin. Die TLRs 3, 7, 8 und 9 binden an mikrobielle Nukleinsäuren, einschließlich doppel- und einzelsträngiger RNA von RNA-Viren und DNA der meisten Organismen. Diese TLRs können eigene Nukleinsäuren (die der Wirtszelle) nicht allein aufgrund struktureller Unterschiede unterscheiden, und die Erkennung fremder Nukleinsäuren (die des Erregers)
‼hängt weitgehend von der Lage in der Zelle ab‼. Alle diese Liganden spielen eine wesentliche Rolle in der Mikrobe und die Mikrobe kann sie nicht eliminieren oder verändern, um sich der Entdeckung zu entziehen. Wissenschaftler haben TLRs in niederen Tieren, wie dem Fadenwurm C. elegans, und in Pflanzen gefunden, was darauf hindeutet, dass TLRs einen alten evolutionären Ursprung in der Wirtsabwehr haben. Auch andere Mustererkennungsrezeptoren für Krankheitserreger werden derzeit identifiziert.”
https://www.nature.com/articles/s41590-022-01163-9

“Kariko et al. fanden heraus, dass die m1Ψ-Modifikation der RNA die durch TLR3, TLR7 oder RIG-I vermittelte Entzündungsreaktion dämpft und die Translationseffizienz und biologische Stabilität erhöht.”
“Studien über mRNA-Impfstoffe gegen Krebs zeigen, dass sie TLR4 (Ref. 14), TLR7 (Ref. 15) oder STING16-Signalwege auslösen können, aber solche Kenntnisse fehlen im Zusammenhang mit BNT162b2.”
”Bali Pulendran war bzw. ist Mitglied im External Immunology Network von GSK und in den wissenschaftlichen Beiräten von Sanofi, Medicago, CircBio und Boehringer-Ingelheim. A.D.G. ist Gastwissenschaftler bei Genentech.”
So, so, liebe Sanofi, GlaxoSmithKline und Boehringer-Ingelheim-Bigpharmaknechte… Ihr konntet nichts beobachten und es ist nichts bekannt?! Wenn ich die Zellprobe nach EINEM Tag entnehme (“Tissues were taken at day 1 after immunization. “) und dann gucke, was da über Wochen passiert. Nunja… Ich zweifle. Oh übrigens liebe Autoren, bevor ich weiter mache:

“Die Makrophagen-Cluster C2 und C15 wurden mit Hilfe von Genen neu eingebettet, die eine epigenetisch umgestaltete Monozytenpopulation charakterisieren, die bei Menschen 21 Tage nach der Impfung mit zwei Dosen H5N1/AS03 angereichert wurde.”
Was zur Hölle sind eigentlich epigenetisch umgestaltete Monozyten? Ich mag meine Monozyten nicht umgestaltet.

Auch sehr hübsch dass ihr einräumt, dass die TLR zwar ansprangen aber dann ja angeblich nicht aufgrund der Plörre, sondern unabhängig?
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8132234/#SD1

“Die Booster-Immunisierung führte jedoch zu einer deutlich breiteren angeborenen Reaktion. Neben der Induktion antiviraler Signalwege führte die Booster-Dosis an den Tagen 22-23 zu einer erhöhten Aktivierung dendritischer Zellen und einer Hochregulierung von TLR-Signalen, Monozyten- und Neutrophilen-Modulen, die zuvor nach der Grundimmunisierung vermindert waren (Abbildung 3c).”
Seltsam, dass sowohl dieses Hübsche Preprint in seinem Supplementary 1 die einzelnen TLRs erfasste, als auch folgende Studien:
https://www.nature.com/articles/s41577-021-00526-x/figures/1

“RNA-Sensoren wie Toll-like Rezeptor 7 (TLR7) und MDA5 werden durch die mRNA-Impfstoffe ausgelöst, und TLR9 ist der wichtigste Doppelstrang-DNA-Sensor für den AdV-Impfstoff.”
Es ist also zu erkennen, dass die Aussagen von Bali Pulendran nicht nur der Studie aus 2021 von John R. Teijaro & Donna L. Farber widersprechen, die begeistert davon schwadronierten wie toll eure Plörre funktioniert. Nein. Auch die Holländer um Föhse kamen auf ein anderes Ergebnis:
medrxiv.org/content/10.1101/2021.05.03.21256520v1.full-text

Und die wurden lustiger Weise von der WHO erwähnt:

https://biorxiv.org/content/10.1101/2022.03.16.484616v1.full

https://ul.qucosa.de/api/qucosa%3A14768/attachment/ATT-0/
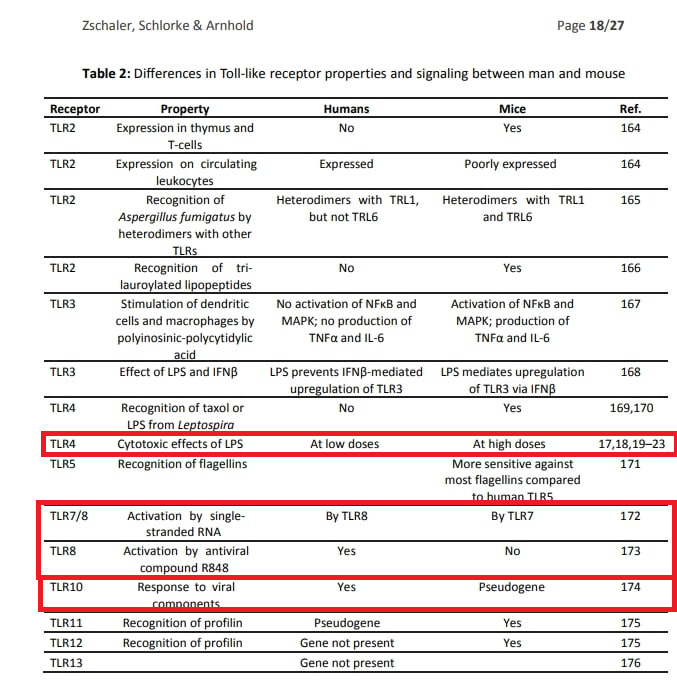
Nochmal zur Erinnerung:

Ich habe eine theoretische Erklärung, die ich jedoch als Laie nicht beweisen kann, da mir dazu einfach die Tiefe an Wissen und Erfahrung mit Labor-Methoden fehlt. Und zudem einfach noch nicht mal ansatzweise ausreichend reproduzierbare Studien existieren. Sie nahmen bewusst Knockoutmäuse anstelle von Primaten. Da, wie aus der Tabelle erkennbar wird, eben der TLR-8 die entscheidende Rolle spielt. Und der arbeitet bei Menschen anders.
Hätte man also wirklich eine Aussage treffen wollen hätte man wohl eher Beobachtungen in beispielsweise Makaken gemacht.
https://www.gatesfoundation.org/about/committed-grants/2014/11/opp1113682
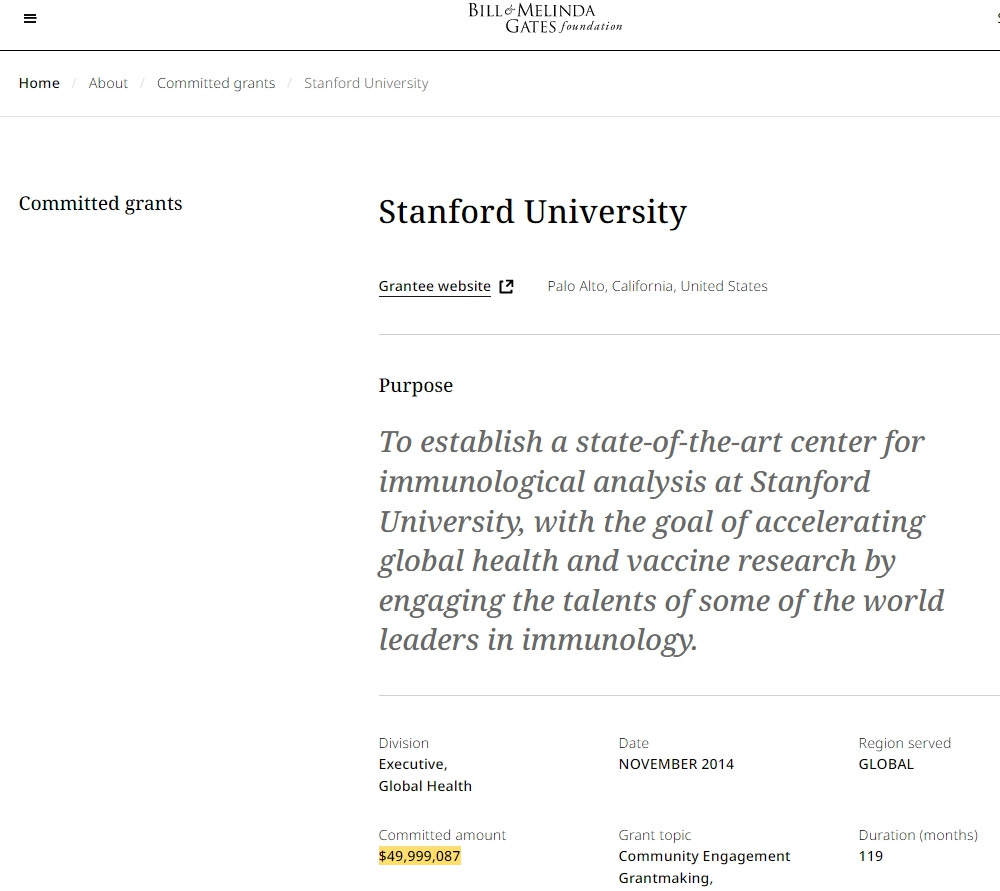
Ich frage mich ja, wie das Funding reinspielen würde, wenn eine in Stanford genehmigte Studie, wie die von Chunfeng Li publiziert wird? Und ob Herr Li eventuell Nähe zur KPC hätte?
Gut, gut. Bis hier hin sind wir also schon mal. Wir wissen jetzt, dass die Toll-Like-Rezeptoren eine essentielle Rolle für die B-Zellen spielen. Genauso natürlich auch für die T-Zellen.
Und damit wäre eine zwingende Kaskade gegeben. Denn der TLR-8 arbeitet niemals unabhängig vom TLR-7 und genauso wenig vom TLR9:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6080926/


Doch wie genau?
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3628816/
”Natürliche Antikörper sind solche, die in erster Linie in der Keimbahn kodiert werden, nicht durch somatische Hypermutation entstehen und Antigenspezifität für eine Reihe von natürlich vorkommenden Epitopen auf der Oberfläche von Mikroorganismen besitzen, darunter Phosphorylcholin, Lysophosphatidylcholin, Phosphatidylcholin und LPS. Diese natürlichen Antikörper haben häufig auch eine Selbstreaktivität und können zu Autoimmunreaktionen beitragen, wie dies beim Rheumafaktor der Fall ist, der zur Bildung von Immunkomplexen und zur Pathogenese der rheumatoiden Arthritis beiträgt. Interessanterweise ist die BCR-Signalübertragung zwar entscheidend für die Entwicklung der B1-B-Zellen, doch reicht sie nicht aus, um die meisten B1-B-Zellen zu aktivieren. Sie können jedoch leicht durch andere Signale aktiviert werden, darunter IL-5, IL-10 und TLR-Agonisten, die ihre Wanderung zu Lymphknoten und Schleimhautstellen auslösen, wo sie sich zu IgM- oder IgA-sezernierenden Zellen differenzieren.”
https://www.frontiersin.org/articles/10.3389/fimmu.2017.00775/full

“B-Zellen sind zu rezeptorvermittelten Reaktionen auf fremde Antigene fähig. Die Erkennung von mikrobieller Nukleinsäure (NA) durch die Toll-like-Rezeptoren (TLRs) 7 und 9 in B-Zellen ist nachgewiesen worden. Endogene NA, die aus geschädigten oder absterbenden Zellen freigesetzt werden, können in bestimmten Zusammenhängen ebenfalls immunogen sein und eine abweichende Aktivierung von B-Zellen hervorrufen. Wenn TLR-gesteuerte B-Zell-Rezeptor (BCR)-aktivierte B-Zellen nicht richtig unter Kontrolle gehalten werden, werden pathologische Autoantikörper gebildet. Es ist auch klar, dass endosomale TLR7/TLR9 in Verbindung mit BCR wirken können. Zusätzlich zur BCR-Signalisierung ist ein Gleichgewicht zwischen TLR7 und TLR9 entscheidend für die Entwicklung der B-Zell-Autoreaktivität. Während TLR9 bei normalen B-Zell-Gedächtnisreaktionen über den BCR eine wichtige Rolle spielt, wurde die Aktivierung von TLR9 mit der Produktion von Autoantikörpern in Verbindung gebracht. Paradoxerweise spielt TLR9 auch eine bekannte Schutzfunktion gegen Autoimmunität, indem es direkt und indirekt die TLR7-vermittelte Autoantikörperproduktion hemmt. Im Folgenden fassen wir die Literatur zusammen, die die Mechanismen zur Förderung pathologischer BCR-aktivierter B-Zellen durch TLR7 und TLR9 unterstützt. Wir konzentrieren uns auf die Literatur zu bekannten Punkten des TLR7/TLR9- und BCR-Crosstalk. Die Daten deuten auch darauf hin, dass der Grad der TLR-Ansprechbarkeit von Veränderungen bestimmter intrinsischer B-Zell-Signalmoleküle abhängt und auch kontextspezifisch ist. Da es sich bei der allogenen hämatopoetischen Stammzelltransplantation um eine Umgebung mit hohem NA- und B-Zell-aktivierendem Faktor handelt, kommen wir zu dem Schluss, dass B-Zell-Studien zur synergistischen TLR-BCR-Signalübertragung bei menschlichen Krankheiten wie der chronischen Graft-versus-Host-Krankheit gerechtfertigt sind.”
Ich hoffe, durch diese eigentlich gut verständlichen Texte wird klar, wieso ich zunächst die TLRs so intensiv diskutieren musste, bevor ich nun auf das Dogma “Antikörper, Antikörper und noch mehr Antikörper” eingehen werde.
Und jetzt lasse ich mal folgenden Satz auf mich wirken:
"Endogene NA (Nukleinsäuren), die von geschädigten oder absterbenden Zellen freigesetzt werden, können in bestimmten Kontexten ebenfalls immunogen sein und eine abnorme Aktivierung von B-Zellen auslösen. Wenn TLR-gesteuerte, durch den B-Zell-Rezeptor (BCR) aktivierte B-Zellen nicht richtig unter Kontrolle gehalten werden, werden pathologische Autoantikörper gebildet.”
Autoantikörper?! Da war doch was…Ich krame mal fix in meinem Schatzkästchen, welches ihr jederzeit unter https://t.me/mspezial durchstöbern könnt und welches permanent wächst.
http://science.org/doi/10.1126/sciimmunol.abp8966

“Hier untersuchten wir eine Kohorte von 48 Personen (Alter 20-86 Jahre), die zwei Dosen eines mRNA-Impfstoffs erhielten und zwei Wochen bis vier Monate später eine Durchbruchsinfektion mit hypoxämischer COVID-19-Pneumonie entwickelten.”
“Unter ihnen hatten zehn (24 %) Autoantikörper, die Typ-I-IFNs neutralisieren (Alter 43-86 Jahre). Acht dieser zehn Patienten hatten Auto-Abs, die sowohl IFN-α2 (alpha) als auch IFN-ω (omega) neutralisierten, während zwei nur IFN-ω neutralisierten. Kein Patient neutralisierte IFN-β (beta).”
“Insgesamt hatten 42 Patienten sowohl keinen B-Zell-Mangel als auch eine normale Antikörperreaktion auf den Impfstoff und wurden daher weiter untersucht.”
Äh…. Liebe Wissenschaftssöldner: Erklärt doch mal, was eine “normale Antikörperreaktion”™ in diesem Fall bedeuten würde? Wollt ihr nicht? - Pah… typisch. Alles muss man selber machen! Dazu also an späterer Stelle mehr.
“Dennoch kann eine hypoxämische COVID-19-Pneumonie bei zuvor gesunden Personen auftreten, die gegen SARS-CoV-2 geimpft sind, was vermutlich auf eine schlechte Antikörperreaktion auf den Impfstoff zurückzuführen ist. Unsere Ergebnisse deuten darauf hin, dass die meisten Fälle von Durchbruchshypoxämie (42 von 48 getesteten) keinen bekannten B-Zell-Mangel aufwiesen und auch eine normale Antikörperreaktion auf den Impfstoff hatten, obwohl keine Proben vor der SARS-CoV-2-Infektion zur Verfügung standen. Darüber hinaus konnten wir zeigen, dass etwa 20 % (10 von 42) dieser Durchbruchsfälle mit normaler Antikörperreaktion auf den Impfstoff auch Auto-Abs tragen, die IFN-α2 und/oder IFN-ω neutralisieren (10 ng/mL bei 7 Patienten und 100 pg/mL bei 3 Patienten). Darüber hinaus neutralisierte das Plasma von 7 der 10 Patienten mit Auto-Abs gegen Typ-I-IFNs effizient SARS-CoV-2 in vitro, während einer eine geringere Neutralisierung gegen den Delta-Stamm aufwies und das Plasma der übrigen 2 Patienten die beiden getesteten Virusstämme suboptimal neutralisierte. Beide Patienten hatten Auto-Abs, die nur 100 pg/ml Typ-I-IFNs neutralisierten. Das Plasma (1/10 verdünnt) von sieben der zehn Personen mit diesen Auto-Abs neutralisierte eine hohe Konzentration (10 ng/mL) sowohl von IFN-α2 als auch von IFN-ω, was damit übereinstimmt, dass ungeimpfte Personen, die solche Auto-Abs tragen, von allen Personen, die eine Kombination von Auto-Abs gegen Typ-I-IFNs tragen, das größte Risiko für eine kritische COVID-19-Infektion haben. Der Anteil der Personen mit hypoxämischer COVID-19 aufgrund der Neutralisierung von sowohl IFN-α2 als auch IFN-ω in der hohen Dosis (10 ng/mL) ist in der hier berichteten Durchbruchskohorte sogar noch höher (7 von 42, 16 %) “
Das nennt man wohl ein weiteres starkes Indiz für zerklumpte Toll-Like-Rezeptoren. Übrigens ist “suboptimal” eine suboptimale Formulierung bei einer solchen Studie. Vielleicht an dieser Stelle mal eine kurze Anmerkung: Wie bereits erwähnt: Zum einen fehlt jede saubere Studienlage, um die Probleme wirklich mit Fakten diskutieren zu können. Es ist also eher ein Puzzlespiel aus bereits bekannten Mechanismen und dem Ausschlussverfahren nach Plausibilitätsprinzipien. Und zum anderen ist - “alles kann, nichts muss”. Bzw: vllt. eher noch: Alles kann, irgendwas wird garantiert passieren. (Ist nur ne Frage der Zeit.) Stellt es euch wirklich wie eine hochkomplexe Zeitbombe vor, deren Countdown man unmöglich vorherbestimmen kann. Man kann nur ahnen, was für drastische Folgen die Detonation haben wird, da man bereits erste Bombenkrater mit einander vergleichen kann. Zurück zum Text:
“Unsere Ergebnisse deuten darauf hin, dass die meisten Fälle von Durchbruchshypoxämie (42 von 48 getesteten) keinen bekannten B-Zell-Mangel aufwiesen und auch eine normale Antikörperreaktion auf den Impfstoff hatten, obwohl keine Proben vor der SARS-CoV-2-Infektion zur Verfügung standen.”
Merkt ihr schon, worauf das hinauslaufen wird und wieso ich immer wieder betone, dass hohe Antikörpertiter ein Nobrainer sind?
Was war gleich der erste Hinweis in diesem Artikel? Richtig: Wie werden B- und T-Zellen denn überhaupt gebildet.
https://jasn.asnjournals.org/content/29/3/741
Na dann werde ich mir nun wohl mal die B-Zell-Aktivierung und Differenzierung im Speziellen ansehen. Wollt ihr noch mit mir weiter reisen?
Im ersten Bild seht ihr eine schöne schematische Darstellung, wie es bei Nieren funktioniert.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2213048/
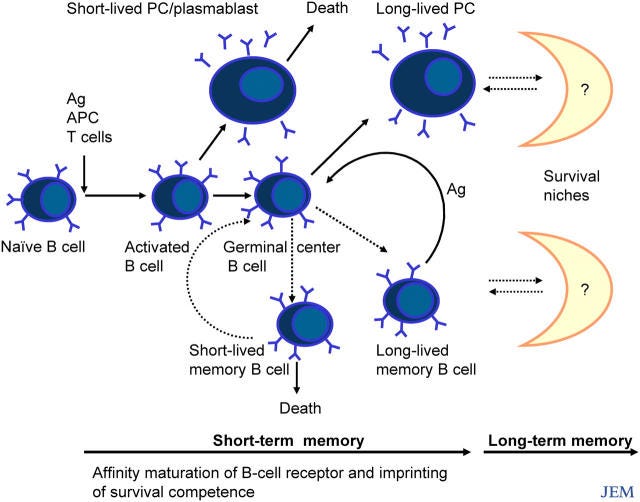
Und jetzt wird es lustig: Da zitiere ich doch nun ausgerechnet Herrn Radbruch, der euch von Antikörpern einen vorbetet, als seien sie das Maß aller Dinge. Herr Radbruch ist einer jener “die Experten”™, die im deutschen Fernsehen gerne mal zitiert werden und vor einem Bücherregal sitzen, damit sie auch voll nach “ich habe Ahnung, vertrau mir”™ aussehen. Zu dumm, dass er selber die Wichtigkeit der Differenzierung von Plasmablasten und B-Zellen betonte.
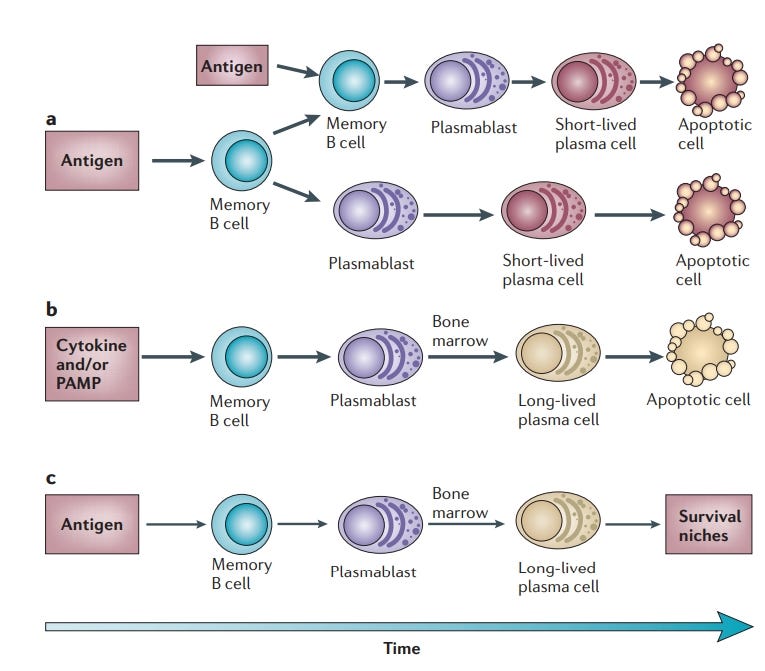
“Plasmazellen sorgen für die humorale Immunität. Traditionell wurden sie hauptsächlich als kurzlebige Endprodukte der B-Zell-Differenzierung betrachtet, die wenig Interesse verdienen. Diese Sichtweise ändert sich jedoch, da wir jetzt wissen, dass Plasmazellen in den entsprechenden Überlebensnischen lange überleben können und dass sie eine unabhängige zelluläre Komponente des immunologischen Gedächtnisses sind. Studien zur Biologie der Plasmazellen enthüllen einen Mechanismus von verblüffender Einfachheit und Eleganz, der das von den Plasmazellen bereitgestellte Gedächtnis auf kürzlich aufgetretene Krankheitserreger fokussiert und gleichzeitig das "Verblassen" des Gedächtnisses für Krankheitserreger, die in der fernen Vergangenheit aufgetreten sind, minimiert. Dieser Mechanismus beruht auf dem Wettbewerb um Überlebensnischen zwischen neu gebildeten Plasmablasten und älteren Plasmazellen.”
Da Herr Radbruch ja so auf die Plasmablasten steht, zeigen wir ihm doch mal, wie es um die nach dem Schuss bestellt ist:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7987037/
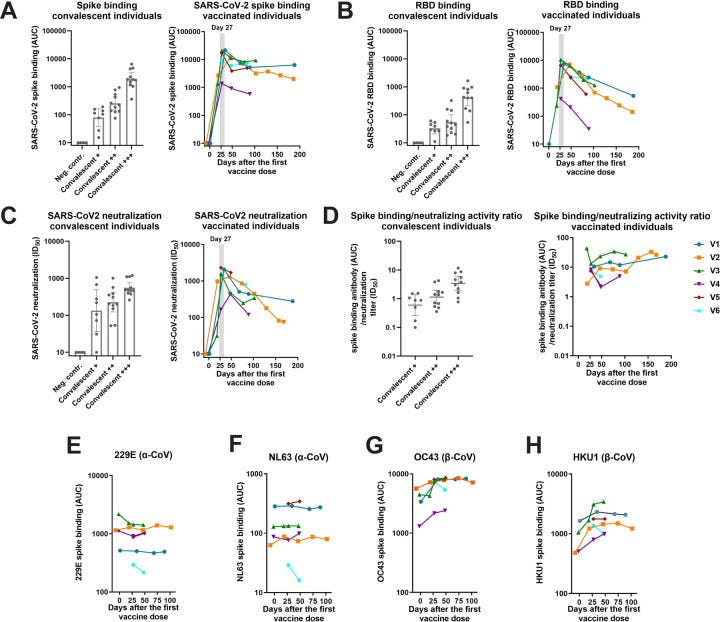
”Darüber hinaus zeigen wir, dass die Mehrheit der isolierten mAbs nicht neutralisierend ist, was das höhere Verhältnis von Bindung zu Neutralisierung widerspiegelt, das im Serum nach der Impfung im Vergleich zur natürlichen Infektion gefunden wurde. Schließlich deuten Daten aus Plasmablasten darauf hin, dass zumindest ein Teil der impfinduzierten Reaktion durch eine bereits bestehende Immunität gegen humane β-Coronaviren beeinflusst wird.”
Wir wissen ja nun längst, dass eine natürliche Infektion der der Impflinge haushoch überlegen ist. Wer sich dazu durch die Studienlage ackern will, es warten mehr als 150 Studien:

Und hier kommt jetzt schon die erste Bombe und es zeigt sich, wieso es so wichtig war, dass ich mir zuvor in vollem Umfang über die CD4 und deren Differenzierung etwas mehr Wissen aneignete:
https://www.frontiersin.org/articles/10.3389/fimmu.2021.731100/full
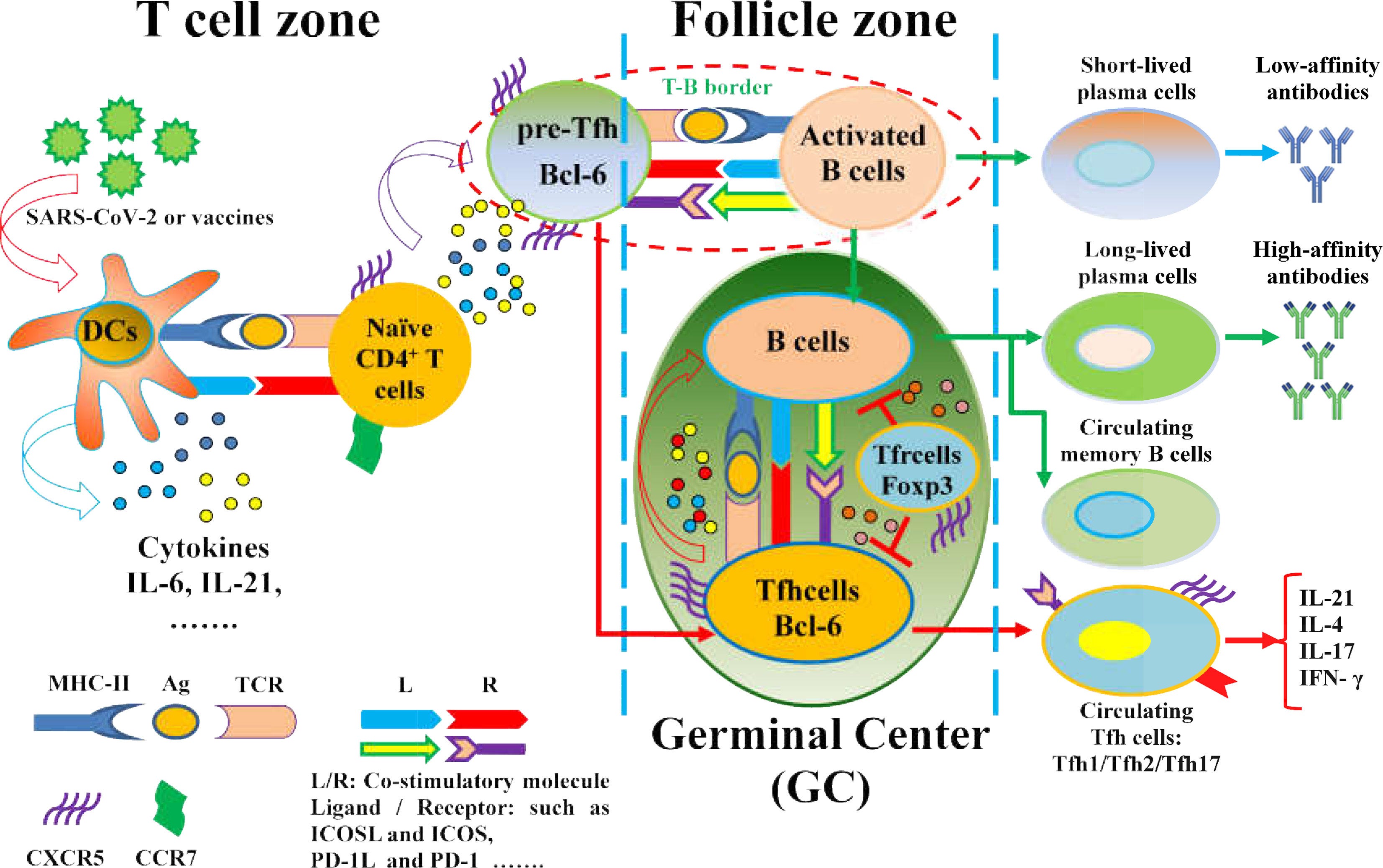
”Tfh-Zellen können B-Zellen durch funktionelle Marker dabei helfen, hochaffine Antikörper, langlebige Plasmazellen und Gedächtnis-B-Zellen zu bilden. Die Marker der Tfh-Zellen sind wichtig, um Tfh-Zellen und ihre verschiedenen Untergruppen im lymphatischen Gewebe und im Blutkreislauf zu identifizieren. Dazu gehören der Chemokinrezeptor CXCR5, der Transkriptionsfaktor Bcl-6, PD-1, der CD40-Ligand (CD40L) und ICOS bei Menschen und Mäusen. Darüber hinaus sind die Phänotypen der Tfh-Zellen mit verschiedenen Stadien der Immunantwort verbunden. In sekundären lymphatischen Organen differenzieren sich naive CD4+T-Zellen in Tfh-Zellen mit einer Hochregulierung von CXCR5 und einer Herabregulierung von CC-Chemokinrezeptor 7 (CCR7), die durch antigenspezifische konventionelle dendritische Zellen (DCs) oder von Monozyten abgeleitete DCs vermittelt werden. Der erhöhte CXCR5 und der verringerte CCR7 tragen zur Migration von Tfh-Zellen in Richtung der mit CXC-Chemokin-Ligand 13 (CXCL13) angereicherten B-lymphoiden Follikel im Keimzentrum (GC) bei. “
Nur blöde, wenn bereits bei der CD4+-Helfer-T-Zelllinie in der Differenzierung falsche Signale getriggert werden? - Ein weiterer Grund, wieso es so wichtig ist, zu zeigen, dass auch die Toll-Like-Rezeptoren vermutlich involviert sind und Kaskaden falsch ausgelöst werden.
https://www.frontiersin.org/articles/10.3389/fimmu.2022.827048/full#f3
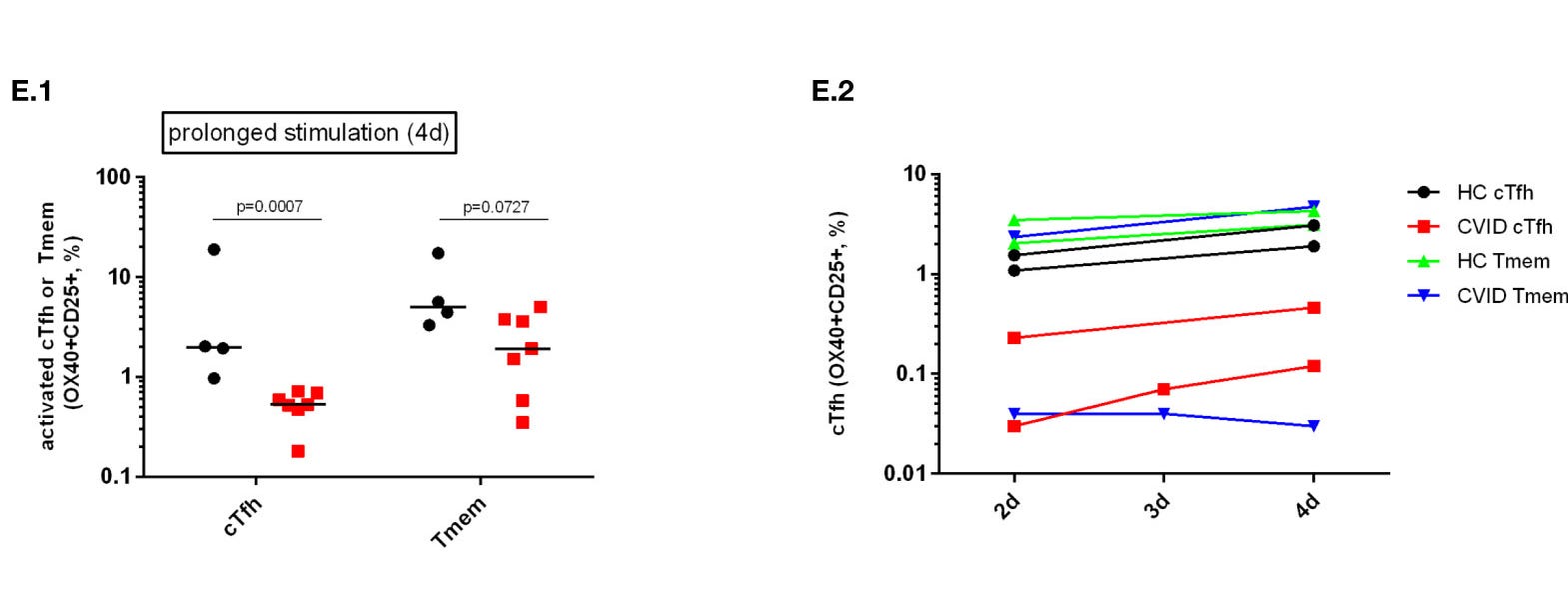
“Unsere Studie zeigt, dass die Aktivierung von cTfh als Reaktion auf die Stimulation mit einem Impfantigen bei CVID gestört ist und dass eine verlängerte Stimulation mit dem Antigen die T-Zell-Aktivierung nicht wiederherstellt, was darauf hindeutet, dass die Aktivierung von CD4-T-Zellen bei CVID nicht nur verzögert ist (Abbildung 3E). Abnormalitäten in der CD4+ T-Zellaktivierung könnten für eine beeinträchtigte antigenspezifische CD4+ Gedächtniszellaktivierung verantwortlich sein, die zu einer Nicht-Ansprechbarkeit auf IgG führt, da die TCR-vermittelte Aktivierung von CD4+ T-Zellen nach SEB-Stimulation bei CVID BNT162b2 IgG-Nonrespondern defekt war. Eine defekte TCR-vermittelte Signalübertragung wurde bereits bei CVID-T-Zellen beschrieben und könnte eine Rolle bei der IgG-Non-Responsiveness spielen, da eine Hemmung der TCR-vermittelten Tfh-Zellaktivierung nachweislich die Antikörperreaktionen und die T-Zell-Hilfe bei der Immunglobulinproduktion in vitro beeinträchtigt. Eine defekte TCR-vermittelte T-Zellaktivierung bei CVID könnte Tfh stärker beeinträchtigen als andere Arten von CD4-positiven T-Zellen wie Tmem. In diesem Zusammenhang wurde beschrieben, dass die Aktivierung von Tfh besonders starke und anhaltende TCR/Liganden-Interaktionen erfordert, Aktivierungsanforderungen, die durch den Kostimulationsdefekt beeinträchtigt werden könnten, von dem zuvor gezeigt wurde, dass er zu einer fehlerhaften TCR-abhängigen T-Zellaktivierung bei CVID führt.”
Also mit anderen Worten: “Hm. Irgendwas ging jetzt zwar höllisch bei der Aktivierung der B-Zellen schief, aber hey, irgendwie ist ja dann doch etwas in den T-Zellen passiert, insbesondere in der CD4. Also kein Grund zur Sorge.”
Wieso eigentlich “C()VID”? Sollte man diese Studie vielleicht nicht finden?
DOI: 10.1038/ni0503-431
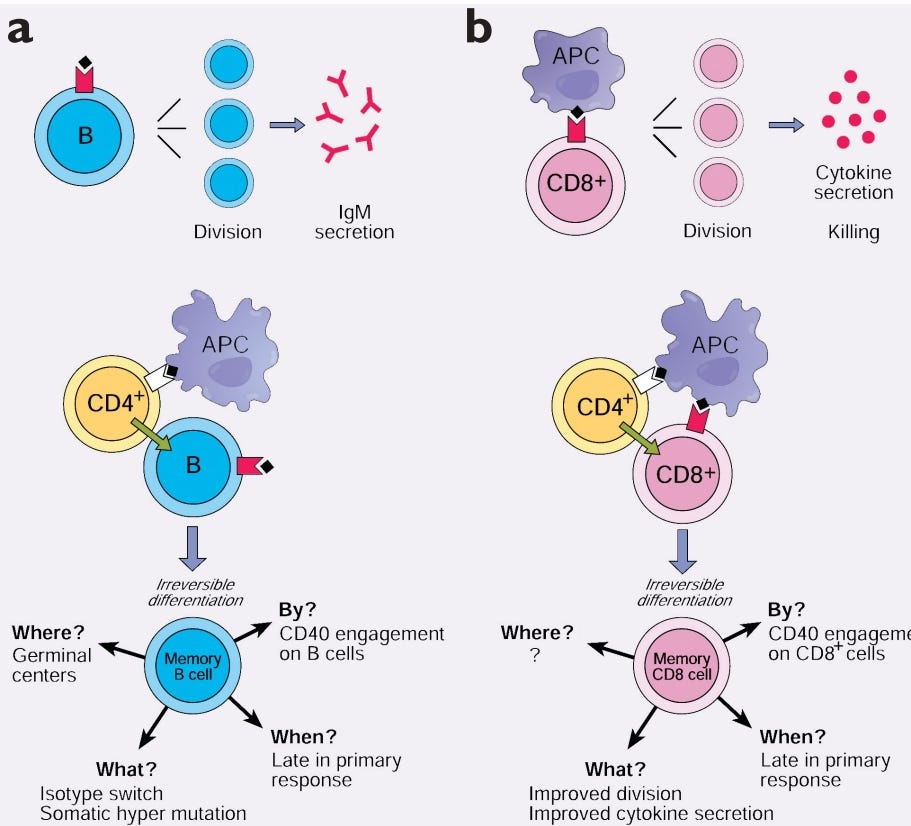
”Die Bildung eines B-Zell-Gedächtnisses erfordert die Hilfe von CD4+ T-Zellen und beinhaltet die Auslösung von CD40 in den Ziel-B-Zellen. Mehrere neuere Studien deuten darauf hin, dass ähnliche Signale an der Bildung von CD8+T-Gedächtnis-Lymphozyten beteiligt sind.”
Hier geht es los: Dieses Paper macht keinen Sinn, wenn ich in Th1 - und Th2 differenziere und den Ort der Rekrutierung berücksichtige. Ich kann nur noch mal betonen, meinem Artikel zu folgen und sich mit der MHC-Class I und II auseinanderzusetzen um jetzt meinen Gedanken zu folgen:
German:
https://genervter.substack.com/p/in-der-hohle-des-lowen?utm_source=%2Fsaved&utm_medium=reader2
English:

https://www.nature.com/articles/s41586-021-03653-6

Zur Erinnerung: Der gesamte Transfektionsschuss wurde vorrangig auf die MHC-Class-I Moleküle des Spikes ausgerichtet und primt sämtliche CD8+, auf die er stößt eben auf diese Epitope, um eine Immunantwort zu formen. Nur dumm, dass die CD8+ flexibel sind und auch in Abwesenheit der CD4+ auf MHC-Class-II switchen können.
Vergleich:
https://www.pnas.org/doi/10.1073/pnas.2116147118


Seltsam dass hier die Furinspaltstelle “QTNSPRRAR” nicht eingebaut wurde, nicht wahr?
https://www.science.org/doi/10.1126/sciimmunol.abg6461
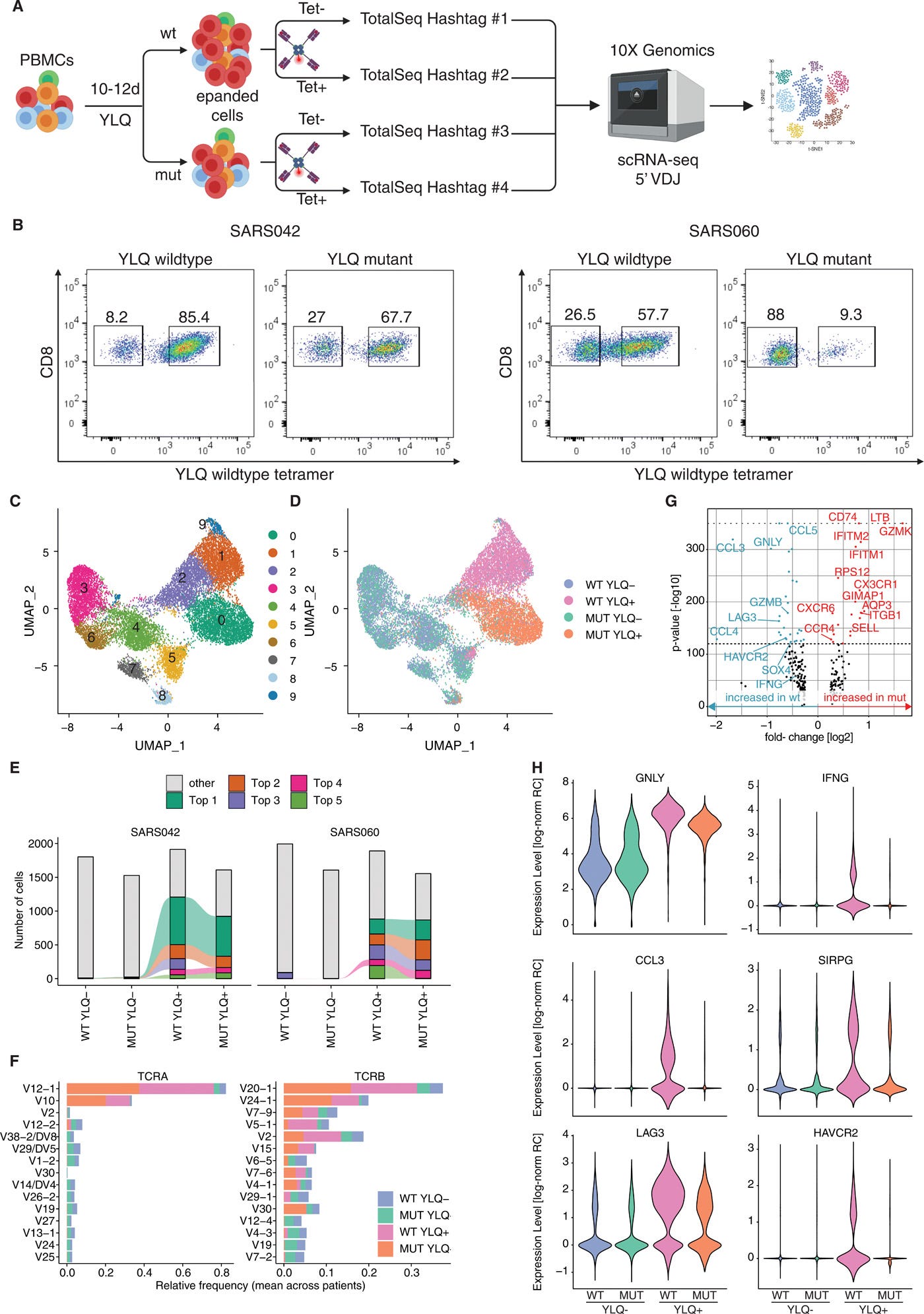
“Die CD8+ T-Zell-Immunität gegen SARS-CoV-2 wurde mit dem Schweregrad von COVID-19 und der Viruskontrolle in Verbindung gebracht. Hier haben wir nach tiefer Sequenzierung von 747 SARS-CoV-2-Virusisolaten nicht-synonyme Mutationen in MHC-I-begrenzten CD8+ T-Zell-Epitopen identifiziert. Die mutierten Peptide wiesen in einem zellfreien In-vitro-Assay eine verringerte oder aufgehobenen MHC-I-Bindung auf. Die verringerte MHC-I-Bindung der mutierten Peptide ging mit einer verringerten Proliferation, IFN-γ-Produktion und zytotoxischen Aktivität von CD8+ T-Zellen einher, die aus HLA-angepassten COVID-19-Patienten isoliert wurden. Die Einzelzell-RNA-Sequenzierung von ex vivo expandierten, tetramer-sortierten CD8+ T-Zellen von COVID-19-Patienten zeigte außerdem qualitative Unterschiede in der Transkriptionsantwort auf mutierte Peptide. Unsere Ergebnisse unterstreichen die Fähigkeit von SARS-CoV-2, die Überwachung durch CD8+ T-Zellen durch Punktmutationen in MHC-I-restringierten viralen Epitopen zu unterlaufen.”
“Es wurden viele CTL-Epitope für SARS-COV-2 beschrieben (39). Natürliche CTL-Reaktionen gegen SARS-CoV-2 wurden mit einer breiten Epitoperkennung von durchschnittlich 1,6 CD8+ T-Zellen-Epitopen pro Antigen und HLA-Allel in Verbindung gebracht, was die Frage aufwirft, ob und wie Mutationen in einzelnen Epitopen die Viruskontrolle beeinflussen. Dies könnte von besonderer Bedeutung für SARS-CoV-2-Untereinheiten-Impfstoffe sein, wie z. B. die derzeit verwendeten RNA-Impfstoffe, die nur das S-Gen enthalten und daher Reaktionen gegen eine begrenzte Anzahl von CD8-Epitopen hervorrufen. Zusammenfassend unterstreichen unsere Ergebnisse die Fähigkeit von SARS-CoV-2, adaptive Immunantworten durch sporadisch auftretende Mutationen in MHC-I-Epitopen zu umgehen.”
Na? Klingt das nicht toll?
https://www.science.org/doi/10.1126/sciimmunol.abg6461
“Durch Abwägung der individuellen MHC-Allel-spezifischen SARS-CoV-2-Bindungskapazität mit der Bevölkerungshäufigkeit in 23 Ländern entdecken wir eine starke umgekehrte Korrelation zwischen der vorhergesagten SARS-CoV-2-Peptid-Bindungskapazität der Bevölkerung und der Sterblichkeitsrate. Unsere Berechnungen zeigen, dass Peptide, die von den Strukturproteinen des Virus abgeleitet sind, eine stärkere Assoziation mit der beobachteten Sterblichkeitsrate aufweisen, was die Bedeutung der S-, N-, M- und E-Proteine bei der Steuerung produktiver Immunantworten unterstreicht.”
Und jetzt bitte anschnallen:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6207480/
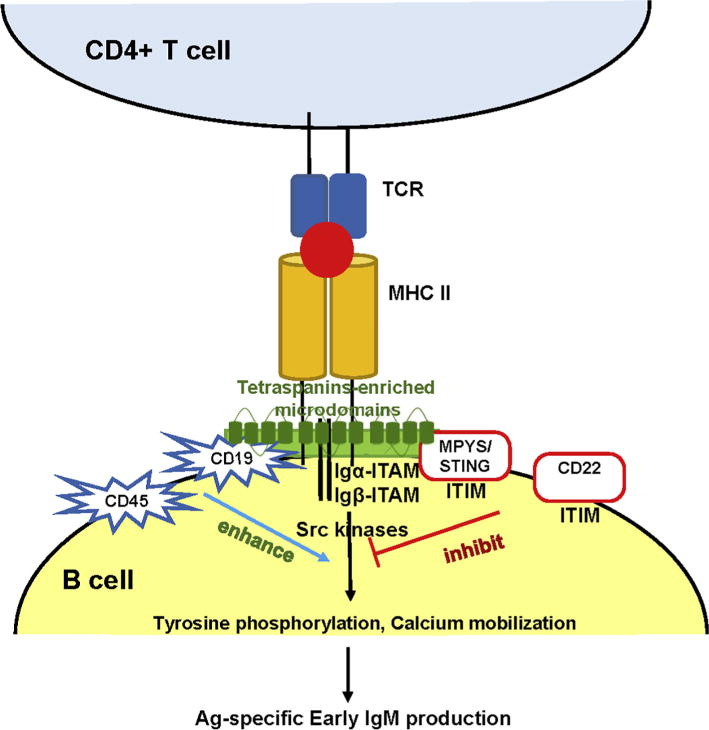
“MHC-Klasse II reguliert die Aktivierung, Proliferation und Differenzierung von B-Zellen während der Interaktion zwischen kognitiven B-Zellen und T-Zellen. Dies ist zum Teil auf die MHC-Klasse-II-Signalübertragung in B-Zellen zurückzuführen. Die Aktivierung der MHC-Klasse II in menschlichen B-Zellen oder "geprimten" B-Zellen der Maus führt zu Tyrosinphosphorylierung, Kalziummobilisierung, AKT-, ERK- und JNK-Aktivierung. Darüber hinaus tötet die Quervernetzung von MHC-Klasse II mit monoklonalen Antikörpern bösartige menschliche B-Zellen ab. Mehrere humanisierte monoklonale Anti-HLA-DR/MHC-Klasse-II-Abs wurden in klinische Studien zur Behandlung von Lymphomen/Leukämie und MHC-Klasse-II-exprimierenden Melanomen aufgenommen. Mechanistisch gesehen ist die MHC-Klasse II mit einer Vielzahl von Transmembranproteinen verbunden, darunter die B-Zell-spezifischen Signalproteine CD79a/b, CD19 und eine Gruppe von vier Transmembranproteinen, darunter Tetraspanine und das apoptotische Protein MPYS/STING. Außerdem werden MHC-Klasse-II-Signale in den mit Tetraspaninen angereicherten Mikrodomänen kompartimentiert.”
Aha… Die MHC-Class-II-Moleküle auf der CD4-Oberfläche bestimmen also über die B-Zellen? - Gott sei dank haben wir ja alles auf das MHC-I-Molekühl geprimt… War also ne richtig gute Idee. Fragt sich gerade noch irgendwer, was zur Hölle da für B-Zellen rauskommen und erlebt das als Geisterbahnfahrt, die einfach keinen Sinn ergibt?
https://www.biorxiv.org/content/10.1101/2020.12.26.424449v1.full
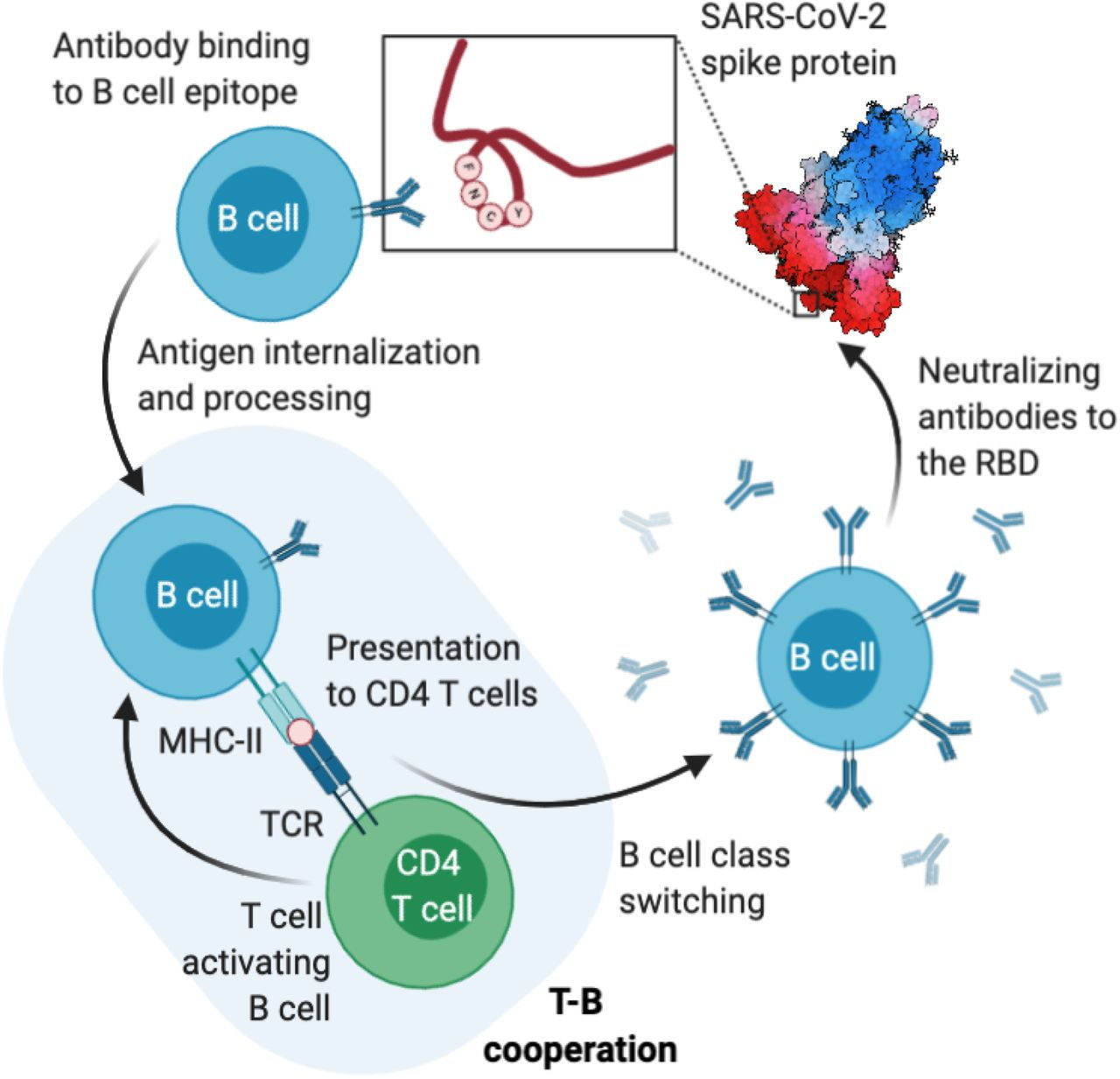
“Die Erzeugung einer Antikörperreaktion erfordert die Zusammenarbeit zwischen einer B-Zelle, die spezifische Antikörpermoleküle produziert, und einer CD4-T-Zelle (Helferzelle), die durch ein Epitop auf demselben Antigen aktiviert wird, das auch von der B-Zelle erkannt wird (T-B-Kooperation). Diese Reaktion findet im Keimzentrum statt. Ausgenommen von dieser Regel sind Reaktionen gegen Kohlenhydrate und Antigene mit sich wiederholenden Motiven, die allein den B-Zell-Antigenrezeptor vernetzen und zur Aktivierung der B-Zellen führen. Vor über 50 Jahren entdeckt, wurde auch deutlich, dass die T-B-Kooperation durch Moleküle des Haupthistokompatibilitätskomplexes der Klasse II (MHC-II) eingeschränkt wird. Die T-B-Kooperation spielt eine Schlüsselrolle bei der Erleichterung und Stärke der Antikörperreaktion, und der Umfang der Antikörperreaktion ist proportional zur Anzahl der Th-Zellen, die von den B-Zellen während der T-B-Kooperation aktiviert werden. Die Bedeutung der T-Zell-Hilfe bei der Aktivierung antigenspezifischer B-Zellen auf Proteinantigene, die die B-Zell-Selektion vorantreiben, wird durch jüngste Experimente unterstrichen, bei denen die Injektion eines Antigenkonjugats (OVA) in Verbindung mit einem Anti-DEC205-Antikörper eine größere Vermehrung von DEC205+ im Vergleich zu DEC205-B-Zellen auslöste, was mit einem T-Helfer-Effekt auf die B-Zell-Aktivierung vereinbar ist.”
“Ein Modell, das auf der Koprozessierung von T- und B-Epitopen beruht, führte auch zu der Vermutung, dass die bevorzugte T-B-Paarung auf topologischer Nähe beruhen könnte, so dass das T-Zell-Epitop während der BCR-vermittelten Internalisierung durch das Paratop des BCR geschützt wird. In der Tat hat eine neuere Studie gezeigt, dass nicht nur die Hilfe der CD4-T-Zellen ein begrenzender Faktor bei der Entwicklung von Antikörpern gegen Pocken (Vacciniavirus) ist, sondern dass es auch eine deterministische Epitopverknüpfung der Spezifitäten bei der T-B-Kooperation gegen dieses virale Pathogen gibt (30). Insgesamt scheint es, dass die T-B-Paarung und die MHC-II-Restriktion Schlüsselereignisse bei der Auswahl der Antikörperreaktion auf Krankheitserreger sind und dass die T-B-Kooperation und die MHC-II-Restriktion Schlüsselereignisse bei der Erzeugung einer adaptiven Antikörperreaktion sind.”
Hm…. Warum dieses kleine Preprint wohl seit 2020 eben als Preprint nicht peer reviewed geführt ist? Gott sei dank sind die Boardmemebers der namenhaftesten Journals ja niemals nie nicht korrupt.
Kleiner Funfact zur MHC-Class-II und den CD8-T-Zellen:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5077698/

“Hier zeigen wir das Vorhandensein von CD8+ T-Zell-Reaktionen mit antiviralen Eigenschaften in einer kleinen Untergruppe von HIV-infizierten Personen, die auf humane Leukozytenantigene (HLA) der Klasse II beschränkt sind. Bei diesen Personen zeigte die Analyse des T-Zell-Rezeptors β (TCRβ), dass CD8+ T-Zellen der Klasse II eine klonale Expansion durchliefen und HIV-infizierte Zellen abtöteten. In einem Fall machten diese Zellen 12 % der zirkulierenden CD8+ T-Zellen aus, und die TCRα-Analyse ergab zwei unterschiedliche koexprimierte TCRα-Ketten, von denen nur eine zur Bindung des Klasse-II-HLA-Peptidkomplexes beiträgt. Diese Daten deuten darauf hin, dass es bei einer chronischen Virusinfektion des Menschen eine Klasse-II-beschränkte Reaktion der CD8+ T-Zellen geben kann, die möglicherweise zur Immunkontrolle beiträgt.”
Das erinnert mich daran, was mir ein sehr erfahrener Arzt, der mich in diese ganze Thematik führte und mich neugierig machte, mehr verstehen zu wollen, einmal bei einer Lernfrage antwortete: “Es gibt nichts in der Biologie, was es nicht gibt. Und meist kommt etwas anderes bei den Experimenten raus, als man erwartet hat.” Danke an dieser Stelle, lieber Herr Passer.
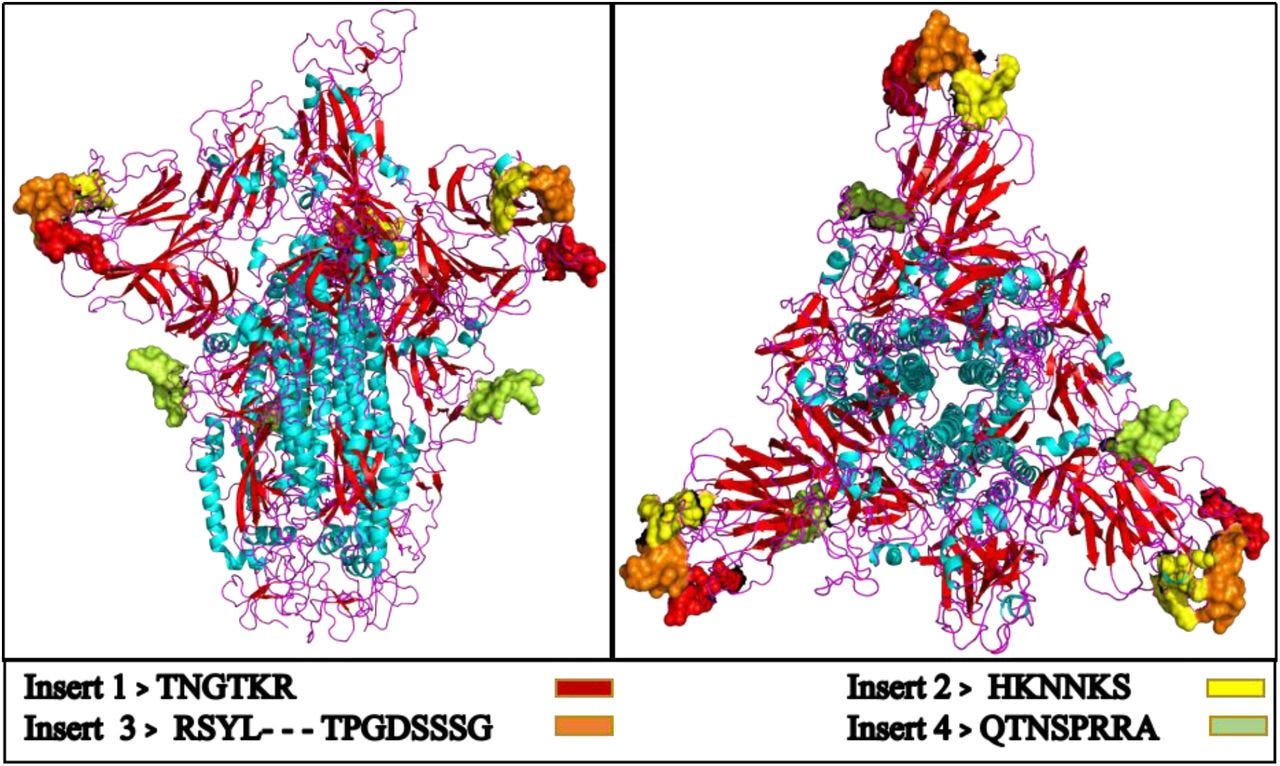

Und hier der Funfact:
“Dieser Artikel zeigt, wie 16 Fragmente (Env Pol- und Integrase-Gene) aus verschiedenen Stämmen der Retroviren HIV1, HIV2 und SIV, die sowohl diversifiziert als auch sehr jung sind, einen hohen Prozentsatz an Homologie mit Teilen des Genoms von COVID_19 aufweisen. Außerdem besteht jedes dieser Elemente aus 18 oder mehr Nukleotiden und könnte daher eine Funktion haben.”
Und jetzt ratet mal, was mit dabei ist: GAG (darum die zitierte MHC-Class-2-CD8+-T-Zell-Studie), GP120 und GP41…. Antikörper gegen HIV?
Doch zurück zu dem Antikörperproblem. Kommen wir nun also final an den Punkt, wie Plasma-Zellen aus den B-zellen entstehen, welche ja die Antikörper produzieren:
Da ich es selbst nicht besser erklären kann, hier noch ein Lernvideo dazu:

”Antigenspezifische B-Zellen teilen sich nach einer Infektion oder Impfung in Antikörper-sezernierende Zellen (ASC) und Gedächtnis-B-Zellen auf. ASC oder Plasmablasten sind beim Menschen umfassend untersucht worden, aber über B-Zellen, die aktiviert werden, sich aber nicht zu frühen Plasmablasten differenzieren, ist weniger bekannt. Hier definieren wir den Phänotyp und das Transkriptionsprogramm einer antigenspezifischen B-Zell-Untergruppe, die wir als aktivierte B-Zellen (ABC) bezeichnen und die sich von den ASCs unterscheidet und zur Gedächtnis-B-Zelllinie gehört. ABCs wurden beim Menschen nach einer Infektion mit dem Ebola- oder Influenzavirus und auch nach einer Impfung nachgewiesen. Durch die gleichzeitige Analyse von antigenspezifischen ASC und ABC in menschlichem Blut nach einer Grippeimpfung haben wir die klonale Überlappung und das Ausmaß der somatischen Hypermutation (SHM) in den ASC- (Effektor) und ABC- (Gedächtnis) Linien untersucht. Die longitudinale Verfolgung der durch die Impfung induzierten HA-spezifischen Klone ergab eine minimale Zunahme der SHM im Laufe der Zeit, was darauf hindeutet, dass eine wiederholte jährliche Immunisierung die Qualität der Influenza-spezifischen Antikörper nur begrenzt verbessern kann.”
Die Studie ist sehr lesenswert.
Wie bereits zitiert, bilden die Transfektionsschüsse ausschließlich Antikörper gegen bestimmte Epitope der MHC-Class-I des Spike-Proteins. Wieso funktionierte das bei HIV nicht?:
https://www.sciencedaily.com/releases/2009/04/090422151832.htm#:~:text=effectively%20become%20stalled.-,The%20reason%3F,they%20are%20meant%20to%20target.
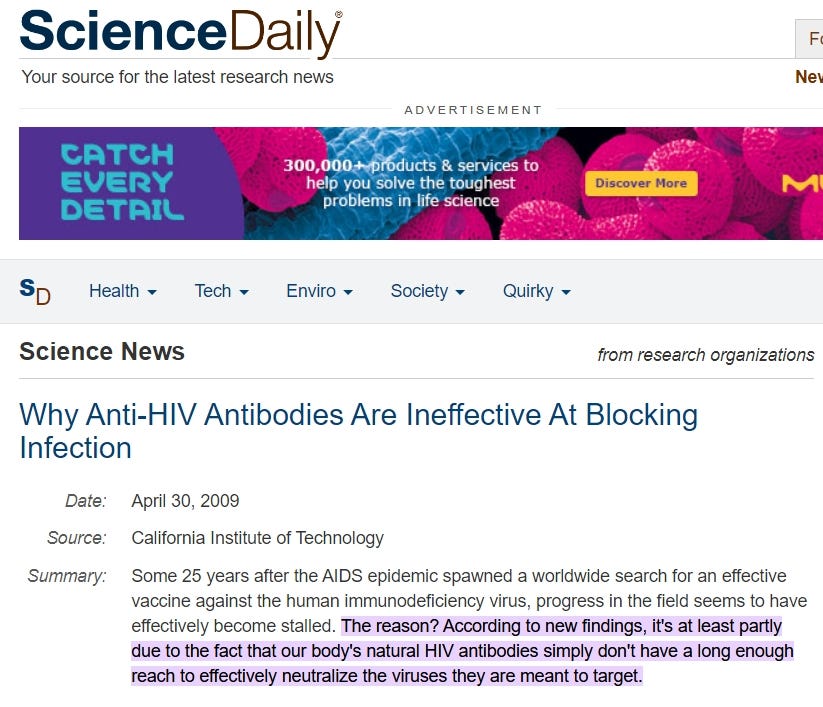
“Der Grund dafür? Neuen Erkenntnissen zufolge liegt es zumindest teilweise daran, dass die körpereigenen HIV-Antikörper einfach nicht weit genug reichen, um die Viren, gegen die sie gerichtet sind, wirksam zu neutralisieren.”
Kommt euch das bekannt vor?
Nachtrag 31.07.2022 (vielen Dank an @Andromaque19841)
Schauen wir uns doch mal noch fix die B-Zell-Subsets bei schweren Krankheitsverläufen an, da ist nämlich gerade eine hübsche Studie erschienen:
https://ncbi.nlm.nih.gov/pmc/articles/PMC9309264/
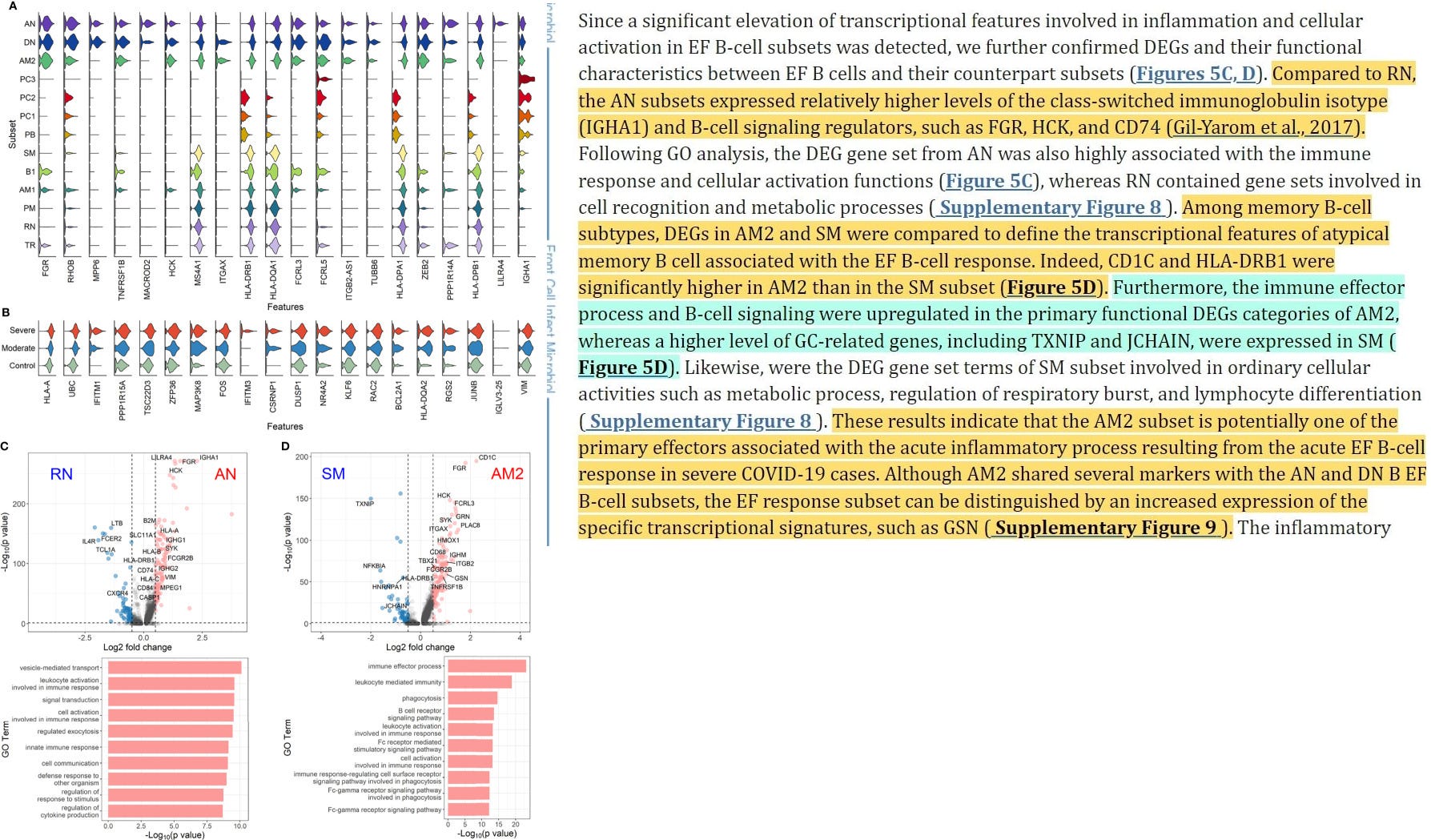
”Im Vergleich zu RN exprimierten die AN-Untergruppen relativ höhere Werte des klassengeschalteten Immunglobulin-Isotyps (IGHA1) und B-Zell-Signalregulatoren wie FGR, HCK und CD74 (Gil-Yarom et al., 2017).

Nach der GO-Analyse war der DEG-Gensatz von AN auch in hohem Maße mit der Immunantwort und zellulären Aktivierungsfunktionen verbunden, während RN Gensätze enthielt, die an der Zellerkennung und an Stoffwechselprozessen beteiligt sind (ergänzende Abbildung 8). Unter den Subtypen der B-Gedächtniszellen wurden die DEGs in AM2 und SM verglichen, um die transkriptionellen Merkmale der atypischen B-Gedächtniszellen zu definieren, die mit der EF-B-Zellantwort verbunden sind. In der Tat waren CD1C und HLA-DRB1 in AM2 signifikant höher als in der SM-Untergruppe (Abbildung 5D). Darüber hinaus waren der Immuneffektorprozess und die B-Zell-Signalübertragung in den primären funktionellen DEG-Kategorien von AM2 hochreguliert, während GC-verwandte Gene, einschließlich TXNIP und JCHAIN, in SM in höherem Maße exprimiert wurden (Abbildung 5D). Ebenso waren die DEG-Gene der SM-Untergruppe an gewöhnlichen zellulären Aktivitäten wie Stoffwechselprozessen, der Regulierung der Atmungsaktivität und der Lymphozytendifferenzierung beteiligt (ergänzende Abbildung 8). Diese Ergebnisse deuten darauf hin, dass die AM2-Untergruppe möglicherweise einer der primären Effektoren ist, die mit dem akuten Entzündungsprozess verbunden sind, der aus der akuten EF-B-Zell-Reaktion in schweren COVID-19-Fällen resultiert. Obwohl AM2 mehrere Marker mit den B-Zell-Untergruppen AN und DN B EF gemeinsam hat, kann die EF-Antwort-Untergruppe durch eine erhöhte Expression der spezifischen Transkriptionssignaturen, wie GSN, unterschieden werden (siehe ergänzende Abbildung 9).

Die entzündlichen Funktionen von AM2 wurden weiter charakterisiert, indem die Anreicherung von Genen mit immunbezogenen Begriffen in 13 klassifizierten B-Zell-Untergruppen berechnet und verglichen wurde. Hier zeigten AM2-Zellen signifikant höhere Transkriptionsprofile für biologische Adhäsion, Zytokinproduktion, B-Zell-Aktivierung, Leukozyten-vermittelte Immunität und angeborene Immunantwort (Abbildung 6). Gleichzeitig war die funktionelle Kategorie der Autoimmunantikörper-Positivität bei AM2 im Vergleich zu anderen B-Zell-Untergruppen relativ höher, außer bei Plasmazellen, PC1 und PC2. Die Analyse des Gensatzanreicherungs-Scores, der speziell in der aberranten Plasmazellpopulation (PC2) erhöht war, zeigte drei entzündliche Signaturkategorien, die signifikant höher waren als in den anderen Plasmazelluntergruppen (Komplementaktivierung, Immunglobulinkomplex und Phagozytoseerkennung) (ergänzende Abbildung 10).

Dies deutet darauf hin, dass die entzündliche Untergruppe PC2 (Plasmazellen subset 2 → ungewöhnliche Abkürzung) eine der primären Zellen sein könnte, die mit dem schweren Krankheitsverlauf bei COVID-19 in Verbindung gebracht werden.”
Na bitte. Das hört sich doch schon mal hübsch an! Antikörper, Antikörper, Antikörper. Aber es wird noch besser. Darum wollte ich diesen Nachtrag noch einbauen, da auch hier die Autoantikörper bewusst betont werden:

“Trotz der massiven weltweiten Bemühungen, die pathogenen Mechanismen von COVID-19 zu identifizieren, sind die primären Verursacher des schweren Lungenversagens, das zu ARDS und tödlichem Ausgang führt, nach wie vor nicht zu erkennen.”
Dazu hätte ich ja jetzt mal nen kurzen Zwischenruf zu machen: Was, wenn SC-2 gar nicht in erster Linie auf die Lungen geht, liebe Autoren? Bei all den Mechanismen, die sich im Spike erkennen lassen, geht es viel mehr um extreme Gewebeschädigungen, neuronale Störungen und Autoimmunerkrankungen. Aber gut. Hauptsache der Narrativ bleibt aufrecht erhalten und die Labor”these” möglichst unausgesprochen, richtig? → Es wäre schließlich niemals nie nicht so, dass eventuell die unterlassene Frühbehandlung + Behandlung mit Remdesivir und viel zu verfrühte künstliche Beatmung die respiratorischen Schäden verursacht haben könnten. Es muss SC-2 gewesen sein.
“Eine dysregulierte Makrophagenreaktion und ein Makrophagenaktivierungssyndrom wurden jedoch als mögliche Ursache für eine schwere COVID-19-Progression identifiziert (Merad und Martin, 2020). Es gibt zahlreiche Belege für die Annahme, dass abweichende Immunreaktionen auf humane Coronaviren durch eine Dysregulation der angeborenen Reaktion, einschließlich der Interferone vom Typ I und III, sowie durch eine abweichende adaptive Immunreaktion gegen das eindringende Pathogen gekennzeichnet sind (Wong und Perlman, 2022). Kürzlich berichtete unsere Gruppe, dass eine verstärkte Eosinophilen-vermittelte Entzündung und die anschließende pulmonale Pathogenese durch verstärkte TH2-basierte Immunantworten mit einer erhöhten Bildung von Immunkomplexen und Membranangriffskomplexen in den Atemwegen und dem Gefäßsystem der entzündeten Lungen, insbesondere bei kritischem COVID-19, in Verbindung gebracht wurden (Kim et al., 2021). Darüber hinaus haben frühere wegweisende Studien über die potenzielle pathogene Rolle dysregulierter humoraler Immunantworten berichtet, einschließlich einer offenen EF B-Zell-Aktivierung (Woodruff et al., 2020) und Autoantikörpern (Bastard et al., 2020; Wang et al., 2021), die mit schwerer COVID-19 korrelieren. Dennoch ist der Mechanismus der abnormen humoralen Reaktion, die an der pathogenen Progression von schwerem COVID-19 beteiligt ist, weitgehend unklar. Hier haben wir die phänotypischen Merkmale der B-Zell-Differenzierung mit Hilfe der scRNA-Sequenzierungsanalyse zirkulierender B-Zellen im Detail untersucht, um ein tieferes Verständnis der pathogenen Natur der B-Zell-Reaktionen im Zusammenhang mit der COVID-19-Morbidität zu erlangen. Durch diese Studie können wir einen EF B-Zell-Antwort-Differenzierungsweg (AN → DN → AM2) identifizieren, der mit dem Fortschreiten und dem Schweregrad von COVID-19 verbunden ist. Darüber hinaus identifizierten wir eine abweichende Plasmazelluntergruppe, PC2, als potenziellen Treiber ‼einer sich verschlimmernden humoralen Reaktion.‼”
Oops?! Macht sachen. Es gibt eine sich verschlimmernde humorale Reaktion? Dabei sind doch Antikörper das Maß aller Dinge? Oder etwa doch nicht?
An dieser Stelle muss ich mich erneut für das endlos tiefe Verständnis für Immunologie von Annelise Bocquet bedanken, die hatte dann nämlich direkt den Geistesblitz passend zu dieser Studie noch folgende Reminder, wo man exakt dieses Phänomen beobachten konnte, rauszusuchen. (Und die sind verflucht gruselig.):

“Humorale Immundefekte (HID) sind heterogene Erkrankungen, die von asymptomatischen Formen, die bei selektiven Mängeln der Immunglobuline A (IgA) und IgG-Unterklassen auftreten, bis hin zu schweren Formen der kongenitalen Agammaglobulinämie reichen. Patienten mit IHL leiden häufig an wiederkehrenden oder schweren HNO- oder Atemwegsinfektionen. Diese Patienten können eine Reihe nicht-infektiöser Komplikationen aufweisen, wie z. B. Autoimmunmanifestationen und Enteropathien, die möglicherweise das einzige auffällige klinische Symptom sind. Schwere Formen der HID lassen sich leicht durch die Messung von Gesamt-IgG, IgA und IgM diagnostizieren. Eine Immunglobulin-Ersatztherapie bleibt die Behandlung der Wahl bei diesen Patienten.”
Da hätte ich ja nun noch ne kleine Anmerkung, die ich fix in einer Slideshow abarbeiten werde. Hier müssen zwei fragen gestellt werden: Wieso drehen die B-Zellen bei den schweren Verläufen so frei und wie könnten wohl B-Zellen positiv beeinflusst werden?:
https://pubmed.ncbi.nlm.nih.gov/20373291/

Was könnte wohl ein akuter Vitmain D3-Mangel mit nem schlechten Immunsystem allgemein zu tun haben? Und wurde es seit 2010 besser mit der Nahrungsmittelbranche?
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4754614/

https://royalsocietypublishing.org/doi/10.1098/rsos.201912
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7456194/

Falls ihr euch jemals gefragt habt, wieso Vitamin D3 nicht mehr richtig funktioniert, wenn ihr bereits krank seid: Vitamin D3 muss erst in ein Pro-Hormon konvertiert werden. Und dieser Prozess braucht Zeit und wird bei einer Infektion vermutlich gestört.
Zurück zu Annelises Gedanken:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4420662/

“B-Zell-Gedächtnisdepletion, Hyperimmunoglobulinämie und beeinträchtigte Impfstoffreaktionen sind das Markenzeichen von B-Zell-Störungen bei Erkrankungen durch das humane Immundefizienz-Virus (HIV). Obwohl die B-Zellen nicht das Ziel der HIV-Infektion sind, gibt es Hinweise auf eine Funktionsstörung der B-Zellen, insbesondere der B-Gedächtniszellen, bei HIV-Erkrankungen, die durch andere Zellen oder HIV selbst verursacht werden. “
Muss ich nicht weiter kommentieren. Das dürfte wohl jeder verstehen.
https://pubmed.ncbi.nlm.nih.gov/31221742/

“Störungen der B-Zellen sind ein Markenzeichen der HIV-1-Infektion. Dies äußert sich in einer erhöhten Anzahl erschöpfter CD21neg-Gedächtnis-B-Zellen, die durch
‼eine kontinuierliche antigenspezifische und zufällige Aktivierung‼ angetrieben werden.”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9309264/

“Trotz des Einsatzes von Impfstoffen und Therapeutika gegen die Coronavirus-Pandemie 2019 (COVID-19) stellt diese schwere Erkrankung eine kritische Belastung für die öffentliche Gesundheit dar, während der Pathogenitätsmechanismus nach wie vor nicht klar ist. In jüngster Zeit häufen sich die Hinweise auf eine mögliche Rolle der abweichenden B-Zell-Reaktion und der humoralen Immunität bei der Krankheitsentwicklung, insbesondere bei Hochrisikogruppen.
Wir haben bestätigt, dass sich akute B-Zellen in Plasmazellen differenzieren, insbesondere bei schweren Patienten, möglicherweise durch eine verstärkte extrafollikuläre (EF) Differenzierung. In den schweren Gruppen wies die erhöhte B-Zell-Antwort im Plasma eine größere Vielfalt an B-Zell-Rezeptoren (BCR) sowie höhere Werte an Anti-SARS-CoV-2-Spike-Antikörpern im Plasma auf als in den gemäßigten Fällen, was auf eine robustere und heterogenere Plasmazell-Antwort bei schweren COVID-19-Patienten hindeutet. Eine Trajektorienanalyse identifizierte einen Differenzierungspfad für die EF-B-Zellreaktion von aktiven naiven zu atypischen Gedächtnis-B-Zellen (AM2) sowie die Entstehung einer aberranten Plasmazelluntergruppe (PC2), die mit dem Fortschreiten und dem Schweregrad von COVID-19 in Verbindung gebracht wurde. Die AM2- und PC2-Untergruppen traten in der akuten Phase der schweren Erkrankung vermehrt auf und wiesen mehrere entzündliche Merkmale auf, darunter eine höhere Zytokinexpression bzw. humorale Effektorfunktion. Diese Merkmale unterscheiden sich von anderen B-Zell-Untergruppen, was auf ein pathogenes Potenzial für das Fortschreiten der Krankheit hindeutet.”
https://www.nature.com/articles/s41598-017-17497-6

Liest sich das nicht insgesamt wie eine sehr schöne Nachricht? Ich frage mich nur, ob hier absichtlich und künstlich ein neuer HIV-Subtyp gezüchtet wird, der diesmal NICHT durch einen Virus ausgelöst wird, weil das natürliche Immunsystem - wenn es intakt und gut genährt ist - ihn ohne große Mühe abwehren könnte. Und Ende 2020 war SC2 bereits endemisch. Es wurde jedoch Panik mit den stumpfen und seltendämliche Mutationsnarrativen getrieben. Und da kaum jemand wirklich die genetische Schiene kapiert (mich inbegriffen), war es ein Leichtes diesen Angstporno voranzutreiben. Das ist einfach eine Tatsache. Diesmal wird es an der Transfektionsspritze liegen und an der unterlassenen frühzeitigen Behandlung mit Vitamin D3, Ivermectin, Hydroxychloroquin, Azithromycin usw. (das ist keine ärztliche Empfehlung). Und auch das ganze Gerede über Long-Covid ist Blödsinn hoch zehn. Und ich sage nicht, dass es das nicht gibt. Nein. Ich leugne auch nicht, dass die Betroffenen wirklich leiden. Um Gottes willen! Aber diese Zustände waren schon bei der Grippe gut charakterisiert und sind nicht neu.
Nachtrag ende
https://www.nature.com/articles/s41467-022-29225-4


https://www.nature.com/articles/d41586-021-01696-3

“Delta ist mäßig resistent gegen Impfstoffe, insbesondere bei Personen, die nur eine einzige Dosis erhalten haben. Eine am 22. Mai veröffentlichte Studie von Public Health England ergab, dass eine einzige Dosis des Impfstoffs von AstraZeneca oder Pfizer das Risiko einer Person, an COVID-19-Symptomen zu erkranken, die durch die Delta-Variante verursacht werden, um 33 % reduzierte, verglichen mit 50 % für die Alpha-Variante.”
https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0119298

“Konkurrenz zwischen IgG-, IgM- und IgA-Antiglykan-Antikörpern im Serum
Anti-Glykan-Antikörper sind eine reichlich vorhandene Subpopulation von Serum-Antikörpern mit entscheidenden Funktionen bei vielen Immunprozessen. Veränderungen in den Spiegeln dieser Antikörper können mit dem Ausbruch einer Krankheit, dem Kontakt mit Krankheitserregern oder einer Impfung einhergehen. Infolgedessen besteht großes Interesse an der Nutzung von Anti-Glykan-Antikörpern als Biomarker für viele Krankheiten. Serum enthält eine Mischung von Anti-Glykan-Antikörpern, die dasselbe Antigen erkennen können, und die Konkurrenz um die Bindung kann möglicherweise die Erkennung von Antikörper-Subpopulationen beeinflussen, die für Krankheitsprozesse relevanter sind. Die im Serum am häufigsten vorkommenden Antikörper-Isotypen sind IgG, IgM und IgA, aber es ist nur wenig darüber bekannt, wie diese verschiedenen Isotypen um dasselbe Glykan-Antigen konkurrieren. In dieser Studie haben wir einen Multiplex-Glykan-Microarray-Assay entwickelt und angewandt, um zu untersuchen, wie verschiedene Isotypen von Anti-Glykan-Antikörpern (IgA, IgG und IgM) um gedruckte Glykanantigene konkurrieren. Während IgG- und IgA-Antikörper typischerweise IgM für Peptid- oder Proteinantigene übertreffen, haben wir festgestellt, dass IgM IgG und IgA für viele Glykanantigene übertrifft. Um die Bedeutung dieses Effekts zu veranschaulichen, liefern wir Beweise dafür, dass die IgM-Konkurrenz die unerwartete Beobachtung erklären kann, dass IgG bestimmter Antigenspezifitäten bevorzugt von der Mutter zum Fötus transportiert zu werden scheint. Wir zeigen, dass IgM in mütterlichen Seren mit IgG konkurrieren, was zu niedrigeren IgG-Signalen als erwartet führt. Da Nabelschnurblut nur sehr geringe Mengen an IgM enthält, wirkt sich die Konkurrenz nur auf die mütterlichen IgG-Signale aus, so dass es den Anschein hat, dass bestimmte IgG-Antikörper im Nabelschnurblut höher sind als im entsprechenden mütterlichen Blut. Insgesamt unterstreichen die Ergebnisse die Bedeutung der Konkurrenz für Studien mit Anti-Glykan-Antikörpern.”
https://www.medrxiv.org/content/10.1101/2021.07.31.21261387v7.full-text

“Wir beobachteten Ct-Werte <25 bei 6.253 von 9.347 vollständig geimpften (67 %) und 6.739 von 11.084 (61 %) ungeimpften Personen (Abbildung 1A)” (Steht im PDF)
Na welch Überraschung: Wenn keine IgM-Antikörper gebildet werden und direkt in Konkurrenz mit IgG-Antikörpern stehen… Wer hätte das da nur vermutet?
Und noch ein kleiner Funfact (Vielen Dank an Jonathan Cuey für diesen Hinweis, da noch mal genau hinzugucken):
https://pubmed.ncbi.nlm.nih.gov/19524620/

Die Antikörper konkurrieren nicht nur untereinander um die Rolle IgM und IgA, sondern auch unter bestimmten Umständen - und auch hier haben wir wieder die MHC-Class im Spiel - mit den T-Zellen und können deren Bindung verhindern. Auch dieses Phänomen war bereits bestens bekannt.
Booster, booster an der Wand, wer ist der glücklichste Transfizierte im ganzen Land?
https://www.medrxiv.org/content/10.1101/2022.02.01.22269931v1

“Arbeitskräfte des Gesundheitswesens, die bei ihrem ersten Test positiv getestet wurden, blieben am häufigsten auch bei ihrem zweiten Test positiv, wobei 56 % der zweiten Tests, die über alle Tage 6-10 hinweg durchgeführt wurden, positiv blieben. Alle ersten Tests, die an den Tagen 5-10 durchgeführt wurden, waren positiv.”
“Mehr als 40 % der geimpften Arbeitskräfte des Gesundheitswesens, die sich gut genug fühlten, um zu arbeiten, wiesen beim ersten Test zwischen Tag 5 und 10 immer noch ein positives RAT-Ergebnis auf. Bei geimpften Personen war die Wahrscheinlichkeit eines positiven Ergebnisses an Tag 5, dem ersten Tag, an dem sie zurückkehren konnten, fast dreimal so hoch und die Wahrscheinlichkeit eines positiven Ergebnisses beim ersten RAT-Test insgesamt ∼2x so hoch."
https://www.frontiersin.org/articles/10.3389/fimmu.2022.867716/full

“Hohe Titer von Antikörpern mit niedriger Affinität bei COVID-19-Patienten stehen in Zusammenhang mit dem Schweregrad der Erkrankung”
Na bitte. Das hört sich doch direkt nach der nächsten Erfolgsmeldung an?
https://www.nejm.org/doi/full/10.1056/NEJMc2202092

Und um noch den wichtigsten Punkt zu unterstreichen, dass auch die B-Zellen durch diese Giftbrühe zerschossen werden, beschäftigen wir uns noch kurz mit dem sogenannten ADCC:
https://www.invivogen.com/adcc-adcp

“ADCC und ADCP werden ausgelöst, wenn mehrere IgG-Moleküle gleichzeitig an FcγRs binden. Die Bindung von Antikörper-Antigen-Komplexen an aktivierende und hemmende FcγRs induziert deren Vernetzung und die anschließende Signalübertragung durch Immunrezeptor-Tyrosin-basierte Aktivierungsmotive (ITAMs) bzw. Hemmungsmotive (ITIMs). Zu den zytoplasmatischen Signalen gehören ein Anstieg der intrazellulären Kalziumkonzentration und die durch Calcineurin/Calmodulin vermittelte Dephosphorylierung von NFAT (Nuclear Factor of Activated T Cells), die seine Verlagerung in den Zellkern und die Bindung an die Promotorregionen von ADCC- und ADCP-relevanten Genen ermöglicht.]”
“ADCC: Ein Überschuss an aktiviertem CD16A induziert die Freisetzung zytotoxischer Granula, die das Ziel abtöten.”
https://onlinelibrary.wiley.com/doi/pdf/10.1002/eji.202149470

“Spezifische RBD-gerichtete Serumantikörper induzieren die ADCC-vermittelte Tötung von Zielzellen durch NK-Zellen
Die Antikörper-induzierte Degranulation von NK-Zellen führt nicht nur zur Freisetzung von Zytokinen, sondern auch von Perforin und Granzymen, die die Abtötung der Zielzellen vermitteln. Um festzustellen, ob die Antikörper-vermittelte NK-Zell-Degranulation auch zur Abtötung von Zielzellen führt, die das SARS-CoV-2-Antigen exprimieren, verwendeten wir die Burkitt-Lymphom-Zelllinie Raji, die für ihre Resistenz gegen die NK-Zell-vermittelte Abtötung unter normalen Bedingungen bekannt ist. Wir modifizierten Raji-Zellen, indem wir die Oberflächenexpression der S1-Rezeptor-Bindungsdomäne (RBD) von SARS-CoV-2 induzierten (Abb. 4A). Das Ergebnis war, dass S1 RBD-gerichtete Antikörper, die durch eine SARS-CoV-2-Infektion induziert wurden, an RBD-exprimierende Raji-Zellen banden, während mit Serum von SARS-CoV-2-negativen Kontrollpersonen keine Bindung beobachtet wurde (Abb. 4A). Darüber hinaus löste das Serum von SARS-CoV-2-Resolvern und von BNT162b2-geimpften Personen eine starke CD107a-Expression durch NK-Zellen als Reaktion auf RBD-exprimierende Raji-Zellen und die Abtötung von RBD-exprimierenden Raji-Zellen aus, während dies bei Serum von SARS-CoV-2-negativen Kontrollpersonen nicht der Fall war (Abb. 4B und C). Wie für den Platten-Degranulationstest beschrieben, war die CD16-Downregulation stark negativ mit der CD107a-Expression korreliert (Spearman's rho = -0,83, p-Wert = 7,52 × 10-5, Supporting Information Abb. S2C). Darüber hinaus wurde beim Vergleich der Ergebnisse des Degranulationstests mit dem Tötungstest ebenfalls eine starke Korrelation beobachtet (Spearman's rho = 0,85, p-Wert = 0,0016, siehe Abb. S2D mit ergänzenden Informationen). Sowohl beim plattengebundenen Degranulationstest als auch beim ADCC-Assay mit Raji-Zellen trugen CD56dim CD16bright NK-Zellen am stärksten zur festgestellten NK-Zell-Degranulation bei (siehe Abb. S5). Insgesamt war die Expression von CD107a durch NK-Zellen signifikant mit der NK-Zell-vermittelten Abtötung von Raji-Zellen verbunden (Abb. 4D, Spearman's r = 0,86, p-Wert = 1,82 × 10-6), was zeigt, dass die SARS-CoV-2-Antikörper-abhängige Aktivierung von NK-Zellen zu ADCC führte.”
Nachtrag 2 - 01.08.2022
https://journals.asm.org/doi/10.1128/JVI.00811-17

”Wir dachten uns, wenn wir die CD4-Bindung durch die Hüllantigene verhindern und gleichzeitig die CD4-induzierbaren (CD4i) Schlüsselepitope erhalten könnten, die für die Virusneutralisierung wichtig und reich an ADCC-Epitopen sind, dann könnten wir die Grundlage für ein verbessertes HIV-Env-Antigengerüst schaffen, das in geeigneter Weise für die künftige Präsentation wichtiger breit neutralisierender Epitope und schließlich für die Deletion dominanter, nicht konservierter Antigenköder modifiziert werden könnte. Unsere Konstruktionskriterien waren zweifach, nämlich Verschluss der CD4-Bindungsstelle und Erhaltung der CD4i-NAb-Epitope, die wir beide in früheren Struktur- und Kleintierstudien nachgewiesen habe.“
Hm… Warum sich dieses Paper wohl ebenfalls mit GP120 beschäftigte?:
”Bislang haben rekombinante monomere gp120- oder oligomere/trimere gp140-Glykoproteine in experimentellen Tiermodellen keine breiten und starken neutralisierenden Antikörper hervorgerufen.“
https://onlinelibrary.wiley.com/doi/10.1111/imm.1346

”Eine gleichzeitige mehrfache Erschöpfung der Lymphozyten ist möglich; diese Situation wird hier als Poly-Lymphozyten-Erschöpfung bezeichnet. Eine Mehrfach-Lymphozytenerschöpfung bei ein und demselben Krebs oder Krankheitserreger wäre besonders gefährlich. Da die Herbeiführung einer Poly-Lymphozyten-Erschöpfung in einem Immunsystem für einen Krankheitserreger erhebliche Vorteile mit sich bringt, gibt es Krankheitserreger mit einer entwickelten Fähigkeit zur Herbeiführung einer Poly-Lymphozyten-Erschöpfung. Zu diesen Erregern gehören bestimmte manipulative Viren, Bakterien, Pilze und protozoische Parasiten.”
Nachtrag 2 ende
Fassen wir also noch mal das gesamte Substack kurz zusammen: Die Toll-Like-Rezeptoren werden höchstwahrscheinlich in diverse verrückte Kaskaden verwickelt sein, auch wenn man uns jede Studie dazu, die Licht in diesen Teilaspekt werfen könnte, verweigert und uns mit betrügerischen Studien abspeisen will. Die B-Zellen und sich daraus formenden Plasmablasten produzieren nutzlose RBD-Antikörper in Unmengen, die auch noch zeitgleich mit funktionellen Teilen des HIV-Virus codiert sind. Die Natürlichen Killer (NK) - Zellen rauschen ab und zudem wird die B-Zelle auch noch mit GP120 und GP41, sowie NEF konfrontiert, wenn sie von der CD4+ aktiviert wird, was so eigentlich aus biologischer Sicht gar nicht passieren sollte. Habe ich irgendwas verpasst?

https://www.nature.com/articles/s41467-022-32376-z

https://biorxiv.org/content/10.1101/2022.02.14.480353v1.full

Ok. Jetzt bin ich echt überzeugt. Ich werde Morgen zum nächsten Impfzentrum meiner Qual, äh, Wahl gehen und mich direkt mit fremder RNA transfizieren lassen, um nutzlose Antikörper zu produzieren, bis sie mir zu den Ohren rausquillen.
Antikörper, Antikörper! Ganz, ganz, ganz, ganz viele bitte!


“Wir konnten in den niederländischen Gemeinden nach den Impf- und Auffrischungskampagnen keinen sterblichkeitssenkenden Effekt der Impfung feststellen. In den beiden Zeiträumen mit hoher ungeklärter Übersterblichkeit konnten wir einen signifikanten, die Sterblichkeit erhöhenden Effekt mit einem 4-Sigma-Wert feststellen. Unsere Ergebnisse ergänzen andere neuere Befunde zur Nullwirkung von MRNA-Impfstoffen auf die Gesamtmortalität und fordern weitere Forschung zu diesem Thema.”

“Die statistisch signifikanten und überwältigend positiven kausalen Auswirkungen nach der Einführung des Impfstoffs auf die abhängigen Variablen Gesamttodesfälle und Gesamtfälle pro Million sollten für die politischen Entscheidungsträger höchst beunruhigend sein. Sie deuten auf einen deutlichen Anstieg sowohl der COVID-19-bedingten Fälle als auch der Todesfälle hin, der direkt auf den Einsatz eines Impfstoffs zurückzuführen ist, der der Öffentlichkeit ursprünglich als "Schlüssel zur Wiedererlangung unserer Freiheiten" verkauft wurde. Die Wirkung der Impfstoffe auf die Gesamtzahl der Fälle pro Million und die geringe positive Assoziation mit der Gesamtzahl der Impfungen pro Hundert deutet auf eine begrenzte Wirkung der Impfstoffe auf die Senkung der COVID-19-assoziierten Fälle hin.”
https://xn--gesundheit-sterreich-ebc.at/evidenz/covid-impfung/

“Schutz vor schwerem Verlauf und Tod
Kann die Impfung vor einem schweren Verlauf schützen? Hier ist die Studienlage nur sehr begrenzt aussagekräftig. Es zeigt sich zwar anfangs eine deutliche Impfeffektivität hinsichtlich der Verhinderung COVID-bedingter Hospitalisierung und Tod, die Gesamthospitalisierung und Gesamtmortalität werden aber nicht angegeben. Die Studien dazu waren nur auf einen kurzen Beobachtungszeitraum von zwei bis drei Wochen begrenzt. Wie lange der gemessene Impfschutz anhält, bleibt unklar.”
NARF’ige Grüße, ein genervter Bürger.
[Vielen lieben dank an Dr. TK Cyrus für das Review]
HI there! sorry for my disturbance. I read through your article but I can't the link between TRLs, T cell and B cell activation in autoantibody production... (I'm way too stupid)
Here's what I got from your article:
+ TLRs crosstalk with BCR siganlling
+ covid mRNA vaccination lower TLR4 and TLR7/8 siganlling -> TLR/BCR crosstalk disturbed? (due to TLR7/9 ratio altered) ->produce autoanbodies?
+ EF B activation (due to inhibition of TCR-mediated Tfh cell activation in CVID patients) is induced -> lacks TCR:MHC2 interaction (CTL can't kill autoreactive B cell; no maturation of b cell antibodies) -> atypical b cell (PC2) with high inflammatory profile produced? (life-long survival niche?) -> worsening humoral response (selective defeciency in either IgM or IgA)
+ useless antibody trigger HIV latency?
+B cell Pertubation as a hallmark in HIV infection (may be covid vaccination as well?)
would you mind to point out my mistake? Thx!