

Poly-A-Tail Readenylierung Teil 3
Die Methodik und der hoffentlich finale Klickmoment

In Teil 1 bin ich ja eher allgemein auf das TCTAG-Motiv eingegangen.
Poly-A-Tail Readenylierung

Vielleicht erinnert sich der ein oder andere noch, als ich noch auf Twitter/X war, bevor zum X Mal gecancelt, lag ich mit dem Hauptautoren der Publikation, die ich hier diskutieren will, im Clinch:
In Teil 2 auf die Silencing-Geschichte und wieso hier bereits ein Haufen von Zirkellogik stattfand.

Widmen wir uns in diesem Substack also noch der grundlegenden Problematik der Methodik und wieso das TCTAG-Motiv der Schlüssel zu Tent4 ist und eben nicht, wie von den Autoren suggeriert, Tent 5. Dazu müssen wir jedoch zunächst die Methodik verstehen:
Was machten denn die Autoren überhaupt? Sprich woher kam das TCTAG-Motiv?
https://www.nature.com/articles/s41586-025-08842-1#MOESM1
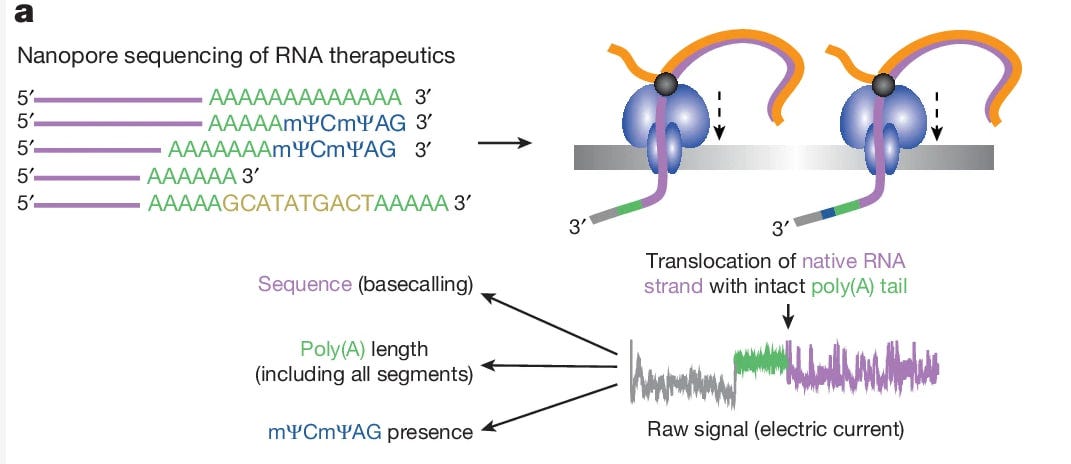
„Schließlich wurde das Vorhandensein eines TCTAG (mΨCmΨAG in der Impfstoff-mRNA) durch schnelle Amplifikation des 3′-Endes der cDNA (3′-RACE) mittels Illumina-Sequenzierung nachgewiesen. Das mΨCmΨAG-Pentamer stellt wahrscheinlich den Rest der Restriktionsenzym-Spaltung der DNA-Vorlage dar. Bemerkenswert ist, dass solche RNAs mit Pentamer-Ende effizient in die Sequenzierbibliothek aufgenommen wurden, obwohl das Protokoll auf der Poly(T)-Splint-Ligation eines Sequenzieradapters an den Poly(A)-Schwanz beruht.“

Es wurde also REAL gemessen und ist der eigentliche - einzig zulässige - Ankerpunkt, für die gesamte Studie.
Doch mein Lieblingsautor, der ja sagt, ich hab nur nicht verstanden, was sie da machten, war extra klug (Deutscher Witz: K L U K):
„Die Analyse der intakten mRNA-1273 mit einer Standard-DRS-Pipeline (Abb. 1a) ergab, dass ein erheblicher Anteil der alignierten Reads den Impfstoff in voller Länge abdeckte (Erweiterte Daten: Abb. 1a, blau), was auf das Potenzial für eine umfassende Analyse von mRNA-Therapeutika hinweist. Die Substitution von Uridin zu mΨ in mRNA-Therapeutika beeinträchtigt jedoch das aufgezeichnete Stromsignal, was zu einer ungenauen Übersetzung in die Sequenz (Basecalling) führt (Erweiterte Daten: Abb. 1b). Daher haben wir einen Subsequenz Dynamic Time Warping (sDTW)-Ansatz entwickelt (Ergänzende Anmerkung 1 und Erweiterte Daten Abb. 1c), der die Identifizierung von doppelt so vielen Sequenzen im Vergleich zur Standard-DRS-Pipeline ermöglicht (Erweiterte Daten Abb. 1a,d-g).“
Sie hatten also akute Schwierigkeiten durch die m1Ψ (auch noch falsch in dem Passus abgekürzt) - Modifikationen mit Nanopore und entwickelten darauf basierend eine neue Methodik zur besseren Erfassung:
Schauen wir uns diese mal noch kurz an:
”Direkte RNA-Sequenzierung von mΨ-haltigen mRNAs Therapeutische mRNA ähnelt normaler mRNA, da sie sowohl eine Cap-Struktur als auch einen Poly(A)-Schwanz aufweist, aber sie wird durch In-vitro-Transkription erzeugt, in der Regel durch T7-Polymerase auf einer DNA-Vorlage. Ein Durchbruch in der Entwicklung von RNA-Therapeutika gelang mit der Entdeckung, dass der Ersatz von Uridin durch N1-Methyl-Pseudouridin (mΨ) die angeborene Immunreaktion verringert und die mRNA-Stabilität erhöht. Obwohl die meisten derzeit synthetisierten mRNA-Therapeutika diese Modifikation enthalten, zeigen Ausnahmen, dass sie nicht wesentlich ist. Das Ersetzen von Uridin durch mΨ in mRNA-Therapeutika bringt zusätzliche Herausforderungen mit sich. Die direkte RNA-Sequenzierung (DRS, Oxford Nanopore Technologies8) basiert auf dem Nachweis von elektrischem Strom beim Durchgang einzelner ssRNA-Moleküle durch eine Proteinpore. Das mΨ in mRNA-Therapeutika stört jedoch das bei der DRS aufgezeichnete Stromsignal und kann zu einer ungenauen Übersetzung in eine Sequenz führen (Basecalling). Unsere Experimente bestätigten, dass mΨ tatsächlich den Basecalling-Prozess beeinflusst (Erweiterte Daten: Abb. 1a,b). Nur 35,74 % der Reads wurden an die Referenzimpfstoffsequenz angeglichen, und diese angeglichenen Reads wiesen nur 74,1 % Identität mit der Referenz auf. Daher ist die Datenanalyse-Pipeline für mΨ modifizierte mRNAs ungeeignet. Um unsere Fähigkeit zu verbessern, die gewünschten Reads zu identifizieren, haben wir einen Subsequence Dynamic Time Warping (sDTW)-Ansatz entwickelt (Erweiterte Daten: Abb. 1c), um Ionenstromsignaturen zu erkennen, die für das 3'-Ende der mRNA-1273 oder BNT162b2 einzigartig sind. Diese Methode ähnelt im Prinzip der gezielten Nanoporen-Sequenzierung10 und ist unabhängig von Basecalling und Mapping. Im Einzelnen wurde die DTAIDistance-Bibliothek (Version 2.3.5) verwendet, um Rohsignale aus der Nanopore-Sequenzierung zu vergleichen. Als Referenzsignale dienten die Reads aus dem DRS-Lauf von rohem mRNA-1273- (read id: df408ab3-7418-4ee4-9a67-92743257b20a) oder BNT162b2-Material (read id: 045d4b9c-9222-425ba7a4-9abb3597f9dd), die eine gute Abdeckung des 3'-Endes entweder der mRNA-1273- oder der BNT162b2-Referenz boten. Die 5000 Datenpunkte, die einen Teil des Poly(A)-Schwanzes (1000 bzw. 500 Datenpunkte für mRNA-1273 und Pfizer) und das 3'-Ende des Transkripts (4000 bzw. 4500 Datenpunkte für mRNA-1273 und Pfizer) abdeckten, wurden für die weitere Verarbeitung ausgewählt (Erweiterte Daten: Abb. 1c). Dieses Fragment der Rohdaten wurde mit dem Savitzky-Golay-Filter (Funktion savgol_filter aus der Scipy-Python-Bibliothek) geglättet, wobei die Fensterlänge auf 51 und die Polynomordnung auf 3 gesetzt wurde, und mit der Funktion zscore aus dem stats-Python-Paket normalisiert. Rohsequenzierungsdaten wurden aus fast5-Dateien mit ONT Fast5 API (Version 4.0.0) gelesen, die ersten 20000 Datenpunkte (die normalerweise das 3'-Ende der sequenzierten RNA abdecken) wurden ausgewählt, dann geglättet und auf die gleiche Weise wie das Referenzsignal normalisiert. Dann wurde die Funktion subsequence_alignment aus der DTAIdistance-Bibliothek verwendet, um eine Region in den Rohdaten zu finden, die am besten mit dem Referenzsignal übereinstimmt. Als Ausgabe wurde für jeden Sequenzierungs-Read die Position der Übereinstimmung und der Distanzwert, der mit der Funktion distance_fast aus der DTAIDistance-Bibliothek berechnet wurde, angegeben. Das Python-Skript für alle beschriebenen Vorgänge ist unter https://github.com/LRBIIMCB/DTW_mRNA-1273 verfügbar. Bei der Anwendung auf Daten, die aus DRS von RNA-Proben aus HEK293T-Zellen, die mit mRNA1273 inkubiert wurden, gewonnen wurden, trennten die mit sDTW berechneten Distanz-Scores die von Impfstoffen stammenden Reads deutlich von den anderen (Erweiterte Daten: Abb. 1d). Auf der Grundlage der Dichteplots wurde entschieden, dass in den nachfolgenden Analysen nur Reads, die auf die mRNA-1273- oder BNT162b2-Referenz gemappt wurden oder nicht gemappt waren und einen mit sDTW berechneten Abstand <10 (im Falle von mRNA-1273) oder <11 (im Falle von BNT162b2) aufwiesen, als von dem jeweiligen Impfstoff stammend betrachtet wurden. Im Vergleich zur alleinigen Kartierung ermöglichte die Kombination von sDTW und Kartierung die Identifizierung doppelt so vieler Sequenzen, sowohl aus der Rohimpfstoffprobe (Erweiterte Daten, Abb. 1e) als auch aus gemischtem RNA-Material von Zellen, die mit dem Impfstoff inkubiert wurden (Erweiterte Daten, Abb. 1f), was die Machbarkeit und das Potenzial dieses Ansatzes bestätigt. Diese Methode war besonders nützlich für die Identifizierung kürzerer, qualitativ minderwertiger 3'-terminaler Reads (Extended Data Abb. 1g), die andernfalls übersehen worden wären. Die mit dem sDTW-Ansatz identifizierten Reads wiesen auch die gleiche Verteilung der Poly(A)-Längen auf wie die mit dem Standard-Mapping-basierten Ansatz identifizierten Reads (Erweiterte Daten: Abb. 2e,g).“
Sie nutzten also eine reine Ionensignatur, welche unmöglich die absolut korrekte Poly-A-Schwanzlänge erfassen kann, auch wenn sie das Gegenteil behaupten.
Und bereits damit zeigt mein Lieblingsautor ein konzeptionelles Missverständnis:
“Die mit dem sDTW-Ansatz identifizierten Reads wiesen auch die gleiche Verteilung der Poly(A)-Längen auf wie die mit dem Standard-Mapping-basierten Ansatz identifizierten Reads (Erweiterte Daten: Abb. 2e,g).”
Diese Aussage ist in sich unzulässig und eine reine Zirkellogik: Die einfache Messung von Ionensignaturen, wie sie bei der Nanopore-Sequenzierung zum Einsatz kommt, kann keine verlässliche Aussage über die genaue Poly-A-Schwanzlänge machen, da diese Methode – unabhängig von der verwendeten Software – auf einem nicht-linearen Signal basiert, das die Länge des Poly-A-Schwanzes nur bedingt widerspiegeln kann. Der Poly(A)-Schwanz erzeugt kein eindeutig interpretierbares elektrisches Signal, da er aus repetitiven A-Nukleotiden besteht. Der Stromfluss ist also nicht-linear und verrauscht ( „nonlinear stochastic system“). Daher kann keine dieser Methoden eine exakte Länge bestimmen. (Siehe Information Dynamics of a Nonlinear Stochastic Nanopore System)
Die zweite Fehlannahme: Readenylierung bedeutet nicht zwingend gleiche Poly-A-Länge, und auch nicht zwingend Aktivität sondern ausschließlich Stabilität!:
Dynamic and widespread control of poly(A) tail length during macrophage activation
”Increases in tail length correlated with mRNA levels regardless of transcriptional activity, and many mRNAs that underwent tail extension encode proteins necessary for immune function and post-transcriptional regulation.”
Poly(A) Tail Length Control Is Caused by Termination of Processive Synthesis (∗)
“The cell controls the length of poly(A) tails in three ways: during synthesis in the nucleus, during shortening in the cytoplasm, and, at least in special cases, during readenylation in the cytoplasm. The first type of control, limitation of poly(A) tail synthesis to a length of approximately 250 nucleotides, can be reconstituted from three purified proteins, poly(A) polymerase, CPSF, and PAB II. The two stimulatory factors, which are also responsible for the primer specificity of the polyadenylation reaction, act by forming a complex that includes poly(A) polymerase and the substrate RNA.”
Habt ihr aufgepasst gehabt, als ich auch den relativen Bezug zur Abundance-Rate kritisierte?:
Enhanced stability and polyadenylation of select mRNAs support rapid thermogenesis in the brown fat of a hibernator (Auch hier geht es um Re-Adenylation)
“The absolute abundance of these transcripts was measured by RT-qPCR in total RNA, and short and long poly(A) RNA fractions (Figure 4—figure supplement 1; Supplementary file 1A) from interbout aroused, late torpor, early arousal, and spring warm animals (n = 3). Two classes of RNA dynamics were apparent; transcripts were either decreased (labeled as Class I) or stabilized (labeled as Class II) during torpor but not newly transcribed.”
mRNA stabilization by poly(A) binding protein is independent of poly(A) and requires translation
Vermutlich hat mein extrakluger Lieblingsautor nur die gängige Lektüre, äh, öh, vergessen?

Doch zurück zu unserem TCTAG + Poly-A und dem Problem:

Bereits in Figure 1e wird das Problem sichtbar: Eine NICHT-Korrelation zwischen ihrem, durch das TCTAG-Motiv + Poly-A, forcierten Signal und dem reinen Poly-A-Signal: Sie behaupten, sie haben CNOT1 (eine Deadenylase) supressiert, um die Deadenylierungsraten zu quantifizieren. Doch wie unschwer erkennbar wird: Es gibt KEINERLEI auch nur im Ansatz plausible Korrelation zwischen TCTAG + Poly-A und dem reinen Poly-A! Ganz im Gegenteil: Nach 72 Stunden sieht man sogar eine verlängerte TCTAG - Poly-A Reaktion (der einzelne blaue Punkt auf der Y-Achse). Bereits damit fällt CNOT1 (welches für TENT5 charakteristisch ist) flach. Denn solange das blaue Signal aufflackert, ist ihr gemessenes Ion-Signal aktiv. So einfach ist die Kiste.
Meine bisherige Argumentation fußte auf einer einfachen Nanopore-Messung. Ich muss zu meiner Schande gestehen, dass ich den Naonopore-Teil erst jetzt im Detail beleuchtet habe und es dadurch nur noch einfacher wird, die gesamten Überlegungen meines Lieblingsautoren, der ja hach so klug wie er ist, kategorisch sein eigenes TCTAG + (plus) Poly-A-Signal ignorierte, ad absurdum zu führen:

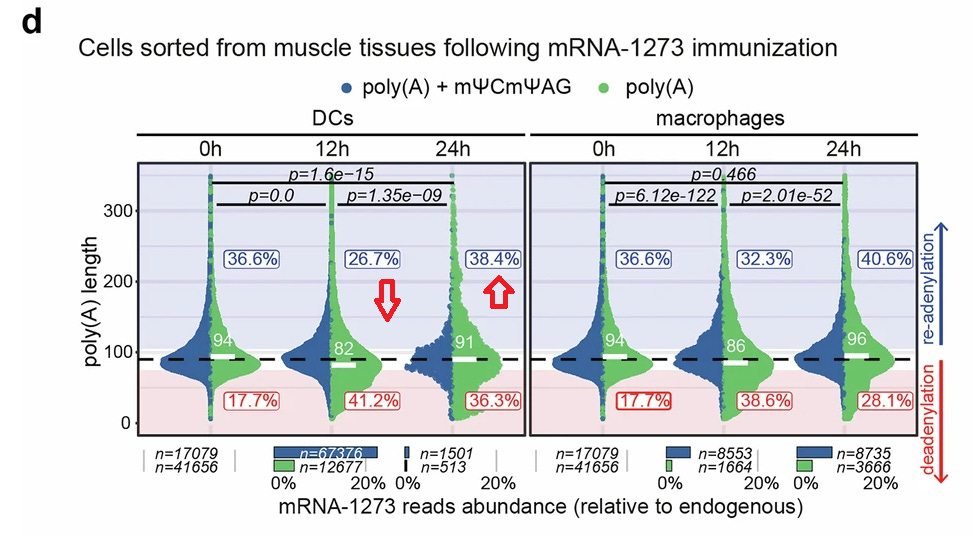
Wir gucken uns die muskelresidenten DCs (Dendritische Zellen) und Makrophagen an, die 12 h und 24 h nach der Impfung mit dem mRNA-1273-Impfstoff aus den Injektionsstellen isoliert wurden. Na so ein Mistekack selbst hier sieht man noch einen leichten Anstieg unseres TCTAG + Poly-A und sogar des geschätzten Poly-A nach 24h im Vergleich zu einer drastischen Abnahme nach 12 h. Und nach 24 Stunden ist der Prozentwert sogar höher als im Ausgangswert bei 0h? Aber, äh?! DCs exprimmieren doch laut meines Lieblingsautoren nur minimal Tent5?!:
”However, DCs express low levels of TENT5A (Fig. 3f,g), raising the question of whether TENT5-mediated stabilization of vaccine mRNA in macrophages affects immune response.”
Ähä… 12 und 24… Wollen wir uns noch mal die frühere Version des Preprints angucken und dagegen stellen?!:
https://www.biorxiv.org/content/10.1101/2022.12.01.518149v2.full
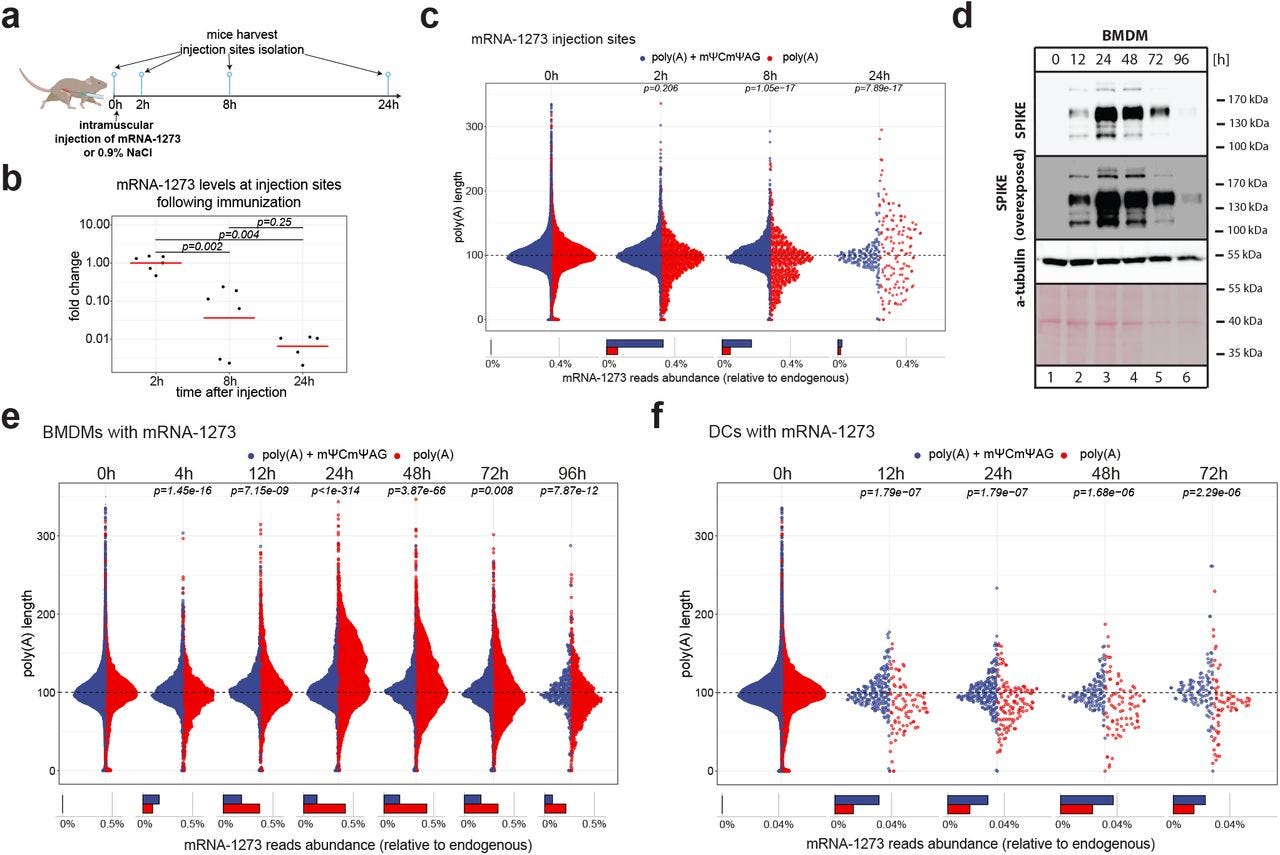
“mRNA-1273 poly(A) tail extension after mΨCmΨAG removal in vivo and in macrophages.
a, Schematic representation of the vaccination experiment
b, Levels of mRNA-1273 at injection sites measured with qPCR 2, 8 and 24 h after injection of mRNA-1273. All values are shown as fold change relative to 2 h timepoint. Red lines represent mean values.
c, mRNA-1273 poly(A) lengths distribution at mRNA-1273 injection sites up to 24 h post-injection. Average distributions for 2-3 replicates are shown. Reads are divided into ones with (blue) and without (red) mΨCmΨAG sequence. Lower panel shows the relative abundance of each group. P.values calculated using Wilcoxon test, p. value adjustment with Benjamini-Hochberg method.
d, Expression of mRNA-1273 in BMDMs up to 96 h after mRNA-1273 delivery. Western Blot on spike protein, α-tubulin as control. Asterisk indicate unspecific band.
e-f, mRNA-1273 poly(A) lengths distribution in BMDMs (e) and DCs (f) treated with vaccine for up to 72 h (DCs) or 96 h (BMDMs). Average distributions for 2 (DCs) or 3 (BMDMs) replicates are shown. Reads are divided into ones with (blue) and without (red) mΨCmΨAG sequence. Lower panel shows the relative abundance of each group. P.values calculated using Wilcoxon test, p. value adjustment with Benjamini-Hochberg method.”
Ach huch! das TCTAG + POLY-A-Signal flackert doch glatt nach 72 Stunden so derbe auf, dass es die gewählte Skala der Autoren sprengte?! Und das ausgerechnet in den DCs. Gut, dass man jetzt in der Nature nur noch 24 Stunden direkt zeigte. Und nochmal muss betont werden: Abundancerate nicht gleich relative (in Bezug/ Kontext) Readenylierungsrate. Und das, obwohl die DCs, wie die Autoren ja selber einräumen, kaum bis kein TENT5 expressieren und zudem die Verteilung der transfizierten Moleküle bei 85 (DCs) zu 115 (mBMDMS) lag.
Wir gehen hier jetzt - um des Friedens willen und um zur eigentlichen Kernaussage zu kommen - davon aus, dass die Autoren in soweit mit ihren Signalmessungen halbwegs richtig lagen, auch wenn wir ja schon geklärt haben, dass die Längenmessung der Poly-A-Schwänze eher eine nette Schätzung ist, denn eine reale Messung:
Dann haben Sie bereits mit der Readenylierung in den DCs ihre eigene TENT5-These widerlegt. Wie bereits Oben erwähnt: Sobald das blaue Signal erscheint, ist ihr TCTAG + POLY-A aktiv. Und das ist in diesem Fall dann eben kein “Unfall” oder ein “Artefakt" sondern exakt dem geschuldet, was sie ja im Methodikteil noch extra stolz verkündet haben. Das ist also nicht meine Interpretation, was ich hier diskutiere, sondern ihre eigenen Daten!
So… Und jetzt bitte Spot on und der endgültige Zwerchfellkitzler. Wir gehen wieder in Figure 5:
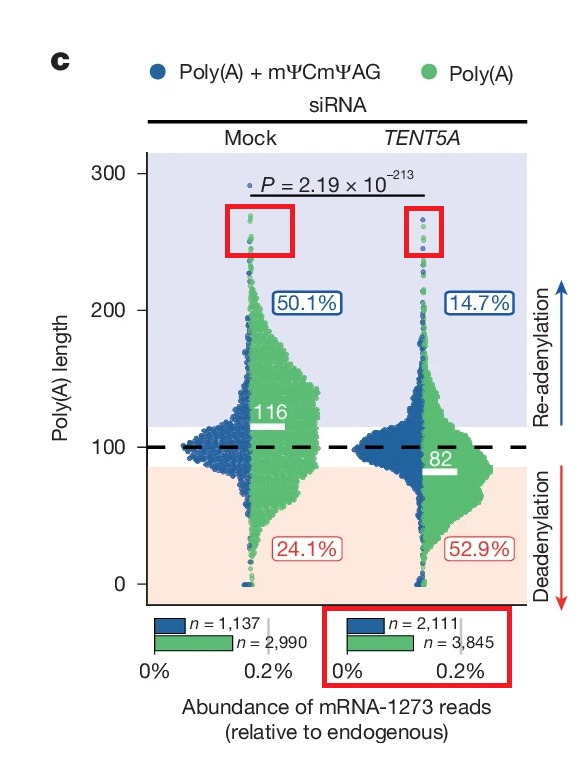
Ei sicher doch: Es war Tent5, weil mit siRNA-Silencing von Tent 5 zwar die Gesamtdeadenylierungsrate einen massiven Sprung erfuhr, aber dafür mein TCTAG + POLY-A-Schwanzsignal und auch Länge (Y-Achse) deutlicher anstieg, sprich sich die Populationen verschoben haben. Das heißt sie setzten nicht die Signalmessung von TCTAG + Poly-A-Schwanz als maßgebendes Signal in den Fokus sondern ihre vermeintlich messbare Poly-A-Schwanzlänge. Wie wir ja schon klärten, ist dies ein unzulässiger Vergleich, da Nanopore zu limitiert dafür ist. Das heißt sie setzten nicht die Signalmessung von TCTAG + Poly-A-Schwanz als maßgebendes Signal in den Fokus sondern ihre vermeintlich messbare Poly-A-Schwanzlänge.

Klar doch. Und das Beste: Während die Gesamtreadzahl NUR bei TENT4a/b-Silencing massiv abnahm, war es natürlich Tent5! Narf! Das kommt dabei rum, wenn man Readenylierung und Deadenylierung mit Poly-A-Länge gleichsetzt! Eigentlich darf man getrost feststellen: Mit der Grafik kann man sich den Arsch abwischen (natürlich bitte vorher ausdrucken) und es hätte einen wissenschaftlichen Mehrwert.
Die Quintessenz zusammengefasst
Die einzige Schlussfolgerung, die sich also aus den essentiellen beiden Beobachtungen ergeben kann, lautet (1. TCTAG + Poly-A ist eine Realmessung und 2. Nanopore kann keine sauberen Daten zu Poly-A-Längen liefern):
Wenn das Ausgangssignal TCTAG + Poly-A, welches eben nicht nur eine künstliche Ionenmessung via Nanopore repräsentiert, im Vergleich zu ihren angeblich konstanten Poly-A-Schwänzen und Längen zunahm, kann es nur Tent4a/b sein:
https://royalsocietypublishing.org/doi/10.1098/rstb.2018.0162
“Da TCTAG ein gemischter Poly-A-Schwanz ist. Siehe: "(d) TENT4A and TENT4B in mixed A/G tailing Based on similarity, it was expected that TENT4A and TENT4B would at least partially functionally overlap, although their functions have mostly been studied separately so far (figure 1). A recent report proposed an interesting not previously described role of both TENT4A and TENT4B [52]. These enzymes were implicated in polyadenylation of protein-coding mRNAs. However, owing to their slightly promiscuous nucleotide specificity, the enzymes occasionally incorporate GMP within the extended poly(A) tails [52]. Figure 2 depicts the process whereby following TENT4A/B activity, deadenylation of the poly(A) tails is executed by CNOT6 L and CNOT7 within the CNOT deadenylating complex. Deadenylation stops on guanine residues owing to the A-preference of CNOT6 L/7, which ultimately results in an increased frequency of guanine residues on the 3′ ends of mRNA with long poly(A) tails [52]. The process is likely more complex owing to the involvement of other factors including poly(A) binding proteins (PABP) that also participate in the adenylation-deadenylation-driven regulation of poly(A) tails [78–80]. While not explicitly stated in the original study [52], the mixed A/G tailing would likely occur in the nucleus given the nuclear localization of TENT4A and TENT4B [49,50,57]. It is, however, also possible that small fractions of TENT4A/B present in the cytoplasm might also participate in mixed A/G tailing. The mechanism of substrate selection and the general impact of mixed A/G tailing by TENT4A and TENT4B on mRNA metabolism remain to be established. Finally, the importance of mixed A/G tailing in different cellular and tissue contexts and at the organismal levels also requires further experimental confirmation."
Es kann also folgerichtig nur ein einziges relevantes Messkriterium übrig bleiben: Dass eine bestimmte TCTAG + Poly-A-Schwanz Aktivitätssteigerung und bei silencing von TENT4a/b eine Gesamtreadminderung gemessen wurde. Da nur dieses Signal tatsächlich eine signifikante Reduktion durch siRNA-silencing von Tent4a/b zeigte. Folgerichtig sind sämtliche Schlussfolgerungen der Autoren wissenschaftlich nicht haltbar.
Die Betriebsblindheit und Logikresistenz meines Lieblingsautoren sollte jedoch vermutlich nicht überraschen: Im Gegensatz zu mir autistischen Nerd der Freude an allen Signalkaskaden, mRNA-Prozessen und Transkriptionsfaktoren, sowie Membraninteraktionen hat, hat mein Lieblingsautor vermutlich sein halbes Leben ausschließlich über seinem TENT5 gehangen. Und nun muss dies ja - bei einem eventuell tatsächlich beobachteten Readenylierungsprozess - zwingend der Treiber sein. Da die modRNA-Kacke ja nicht in den Nukleus kann (weil wegen ist so, auch wenn niemand so wirklich erklären kann, wieso das beispielsweise nicht über Line1 passieren könnte).
Und um jetzt die finalen und endgültig zerstörenden Abschlussfragen zu stellen:
Wie habt ihr exkludiert, dass es zu Stop Codon-Readthroughs kam?:
Pseudouridine-mediated stop codon readthrough in S. cerevisiae is sequence context–independent
Wie habt ihr exkludiert, dass es zu einem POLY-A-Ablesen kam?:
https://pmc.ncbi.nlm.nih.gov/articles/PMC5393183/
Wie habt ihr sichergestellt, dass es nicht zu einem nuklearen Transport kam, durch Poly-A in Poly-K-Übersetzung?
https://pmc.ncbi.nlm.nih.gov/articles/PMC11449308/
Zusammenfassung der Autorenlogik:
👉 Poly(A)-Tails verlängern sich? ➔ "Muss TENT5 gewesen sein!"
👉 TCTAG-Motive bleiben stabil? ➔ "Muss TENT5 gewesen sein!"
👉 CNOT1-Deadenylierungssilencing zeigen keine Korrelation? ➔ "Ähm... trotzdem TENT5!"
👉 TENT4A/B-Silencing verändert TCTAG-Poly(A)-Tails sichtbar? ➔ "Bestimmt TENT5, oder...?"
👉 Stopcodon-Readthrough möglich ➔ "Pssst, darüber reden wir lieber nicht."
Ich hoffe, dass ich mit Teil 3 euch meine ersten beiden Teile noch ein wenig greifbarer erklären konnte. Narfing schönen Nachmittag. Euer Genervter TENT2, äh, Narf5, ach, na ihr wisst schon.