
Die phospholipide Doppelschicht und die MAPkkk/kk/k mit Augenmerk auf RAS(k/n/h)
Die phospholipide Doppelschicht und die MAPkkk/kk/k mit Augenmerk auf RAS(k/n/h)
The good, the bad and the transfection


Wow. Es ist schon wieder ganz schön lange her, dass ich mein letztes Substack schrieb. Darin behandelte ich ja die Micro-RNAs, mögliche Alterationen und die Unbekannten, die N1-methyliertes Pseudouridin (m1ψ) darauf haben kann. Seitdem beschäftigte ich mich jedoch noch intensiver mit den Signalkaskaden, da diese meiner Meinung nach sträflich unterschätzt sind und dort, noch bevor all die Unbekannten der Translation und Proteinexpression zu diskutieren wären, die erste Shitshow schon geschehen wird. Wie immer gilt: Ich bin kein Wissenschaftler und kein Arzt, sondern "nur" ein autistischer Nerd, somit findet ihr hier keine medizinischen Anweisungen oder Behandlungsempfehlungen. Ich lerne auch jedes Mal noch neue Dinge dazu, wenn ich in meinen Substacks das gesammelte neue Material auswerten möchte und muss wesentlich mehr erklären, als sich eigentlich angesammelt hätte. Es wird also wieder ein Höllentrip aus Kaskaden, Crosstalks und Check-and Ballancemechanismen. Interessanter Einschub vom jetzigen Stand dieses Höllenrittes: Das wird das erste Substack sein, an dem ich mehrere Tage arbeite und einige Studien wirklich systemisch mit euch diskutieren muss, um sicher zu gehen, dass ihr meinen Gedanken folgen könnt. Dabei merke ich, dass ich auch viele Studien oft nur überfliege und viele feine Details selber beim tieferen Lesen noch neu dazu lerne. Noch ein kleiner Einschub, der mir gerade auffällt: Genexpression = Genexprimierung (für die deutsche Version wichtig, weil es 2 Termini für die selbe Sache gibt). Worauf dieses Substack hinarbeiten wird, sind unter anderem diese beiden Papers, die ich an späterer Stelle erst diskutieren möchte. Der Screenshot ist erstmal dazu da, um zu zeigen, dass sich meine Fragen nach den Signalkaskaden und Veränderungen an den membrangebundenen Rezeptoren in Funktion logisch ableiten und sich auf vorhandene Papers stützen.

Doch bevor wir nun eintauchen muss ich noch ein Problem ansprechen, welches mir beim Schreiben schmerzlich bewusst wird: Science is a bitch! Während das folgende Paper behauptet, es konnte keine Entzündungsreaktionen beobachten mit der Verwendung VERGLEICHBARER Lipid Nanopartikel,
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8695324/


sagt das Paper von Ndeupen et al., es konnte eben diese Entzündungsreaktionen unter Verwendung der von Acuitas hergestellten Lipidformulierungen für BNT162b2 beobachten:
https://www.cell.com/iscience/fulltext/S2589-0042(21)01450-4

Das Zauberwort ist also "vergleichbar"™/”ähnlich”™ um eine Behauptung aufzustellen, die die eigentliche Frage nicht behandelt. Um es dann abzurunden und endgültig zu verwirren, beschrieben sie die von BNT162b2 genutzten LNPs noch näher. Payed by BioNtech und NIH. Ich gehe an späterer Stelle auf das Ndeupen Paper, das ich gerade zitiere, noch ein. Ein weiterer Aspekt der zu berücksichtigen ist, ist dass die Tiere in der BioNTech bezahlten Studie wenige (4) Stunden später getötet wurden. ("m1ψ-haltige mRNAs wurden in LNP eingekapselt, indem eine wässrige Lösung der mRNA bei einem pH-Wert von 4 schnell mit einer Lösung von in Ethanol gelösten Lipiden vermischt wurde [17]. Sofern nicht anders angegeben, waren die in dieser Studie verwendeten LNP ähnlich zusammengesetzt [‼]wie[‼] die zuvor beschriebenen [17,18], die ein ionisierbares kationisches Lipid (Eigentum von Acuitas), Phosphatidylcholin, Cholesterin und PEG-Lipid enthalten.")
Gehen wir zur Ausgangsproblematik: Die phospholipide Doppelschicht (Eile mit Weile - ein langes Vorwort für das eigentliche Problem)
Beschäftigen wir uns heute also einmal näher mit dem Teil, der als erster von den mod-RNA(simulations)Brühen betroffen sein wird: Die phospholipide Doppelschicht (Plasmamembran / Zellmembran). Die grundlegenden Funktionen lassen sich nicht so ohne weiteres erklären und wer hier noch keinerlei Grundkenntnisse hat, wie Diffusion, Endozytose (Phagozytose, Pinozytose) (umgekehrt Exozytose) und Transportermoleküle (Transferasen/ Carrier) funktionieren, sollte sich unbedingt folgendes Video antun, um die Basisfunktionen zu verstehen wie eine Zelle Stoffe aufnimmt:
Und hier wird es dann auch mal wieder richtig kompliziert (wie sollte es auch anders in meinen Substacks sein), weil das Video zwar die Strukturen richtig erklärt, jedoch die Funktionen in einem Kernpunkt unterschätzt: Die Signalkaskaden werden eben nicht nur durch Hormone ausgelöst sondern durch JEDE phospholipide Interaktion. Was - fängt man an die Mechanismen zu verstehen - auch absolut Sinn ergibt, da jede intrazelluläre Interaktion klare regulatorische Mechanismen benötigt und ja auch inter und extrazelluläre Reaktionen abgeben muss.
Schauen wir uns also zunächst einmal die Membranorganisation an:

https://www.nature.com/articles/s41580-022-00490-x
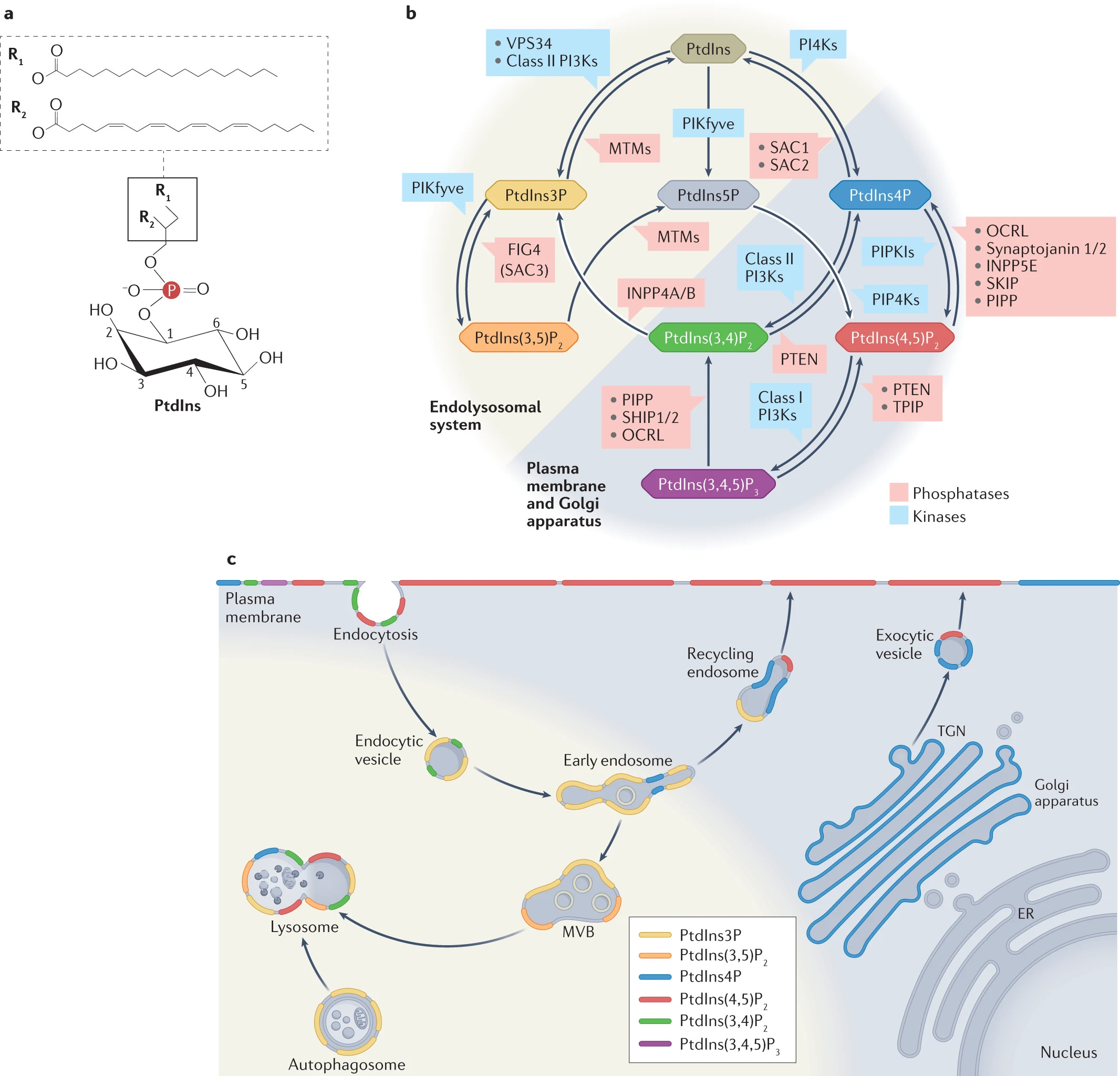
Phosphoinositide als Membranorganisatoren
“Viele Organellen können auch Membrankontaktstellen (MCS) untereinander bilden, um den nichtvesikulären Austausch von Lipiden, Ionen und Metaboliten zu ermöglichen. Schließlich durchlaufen Organellen Fusions- und Spaltungs- sowie Reifungsprozesse, die der physiologischen Reaktion der Zelle auf sich ändernde Umweltbedingungen, wie Hunger oder Stress, zugrunde liegen. Diese Prozesse, die zusammenfassend als "Membrandynamik" bezeichnet werden, erfordern eine fein abgestimmte Regulierung der Interaktion von Proteinen mit Membranen. In den letzten Jahrzehnten hat sich gezeigt, dass Phosphoinositide - d. h. phosphorylierte Derivate des Myo-Inositol-haltigen Membranphospholipids Phosphatidylinositol (PtdIns; Abb. 1a,b) - die Identität der Kompartimentmembranen maßgeblich bestimmen und als räumlich-zeitliche Anhaltspunkte zur Steuerung der Membrandynamik dienen. So fungiert beispielsweise PtdIns-3-Phosphat (PtdIns(3)P) als Markerlipid, das wichtige zytosolische Proteine an frühe Endosomen rekrutiert, um deren Identität zu definieren.”
Zusammenfassend: Entgegen der Annahme, dass die Phospholipide der Membran homogen wären, was ja zu gerne von den Transfektionsnazis suggeriert wird, zeigt sich also, dass sie heterogen sind. Zudem haben sie wichtige Früherkennungsfunktionen, indem sie die Endosome, die in die Zelle gehen, markieren und für die weitere Verarbeitung definieren.

Anschnallen! Es wird jetzt holprig:
“PtdIn wird im ER synthetisiert und über Lipidaustauschproteine an MCSs und durch vesikuläre oder röhrenförmige Träger, die sich entlang des sekretorischen Weges und innerhalb des endolysosomalen Systems bewegen, an andere Organellen abgegeben. Durch reversible Phosphorylierung und Dephosphorylierung des Inositolrings durch Phosphoinositid-Kinasen und -Phosphatasen entstehen sieben verschiedene Phosphoinositid-Spezies. Diese Spezies können je nach enzymatischer Aktivität ineinander umgewandelt werden, was zu einem Stoffwechselnetz mit vielfältigen gegenseitigen Abhängigkeiten führt (Abb. 1b). Phosphoinositide machen weniger als 1 % des gesamten Phospholipidpools der Zellen aus, weisen jedoch aufgrund der engen räumlichen und zeitlichen Kontrolle ihrer Synthese und ihres Umsatzes eine stark lokalisierte subzelluläre Verteilung auf (Abb. 1c). Dies wirkt sich direkt auf die Lokalisierung und Aktivität von zytosolischen Effektorproteinen und membranintegralen Proteinen wie Rezeptoren, Ionenkanälen oder Transportern aus, die mit diesen Lipiden interagieren und eine physiologische Reaktion auslösen (z. B. eine Signalkaskade oder die Bildung eines Proteinkomplexes, der die Membranen umgestalten kann). Phosphoinositid-bindende Domänen von Effektorproteinen binden im Allgemeinen mit geringer Affinität an ihr Ziellipid. Infolgedessen sind für die Funktion von Effektorproteinen eine multivalente, gleichzeitige Erkennung zusätzlicher Faktoren, einschließlich kleiner GTP-bindender Proteine und anderer Lipide, sowie Aviditätseffekte (Anziehungseffekte) erforderlich, die durch das Vorhandensein mehrerer Lipidbindungsstellen oder -domänen auf Effektorproteinen verursacht werden. Die duale, übereinstimmende Erkennung von Lipiden und Proteinen liegt auch der beobachteten Kompartmentspezifität von Lipid-Biosensoren auf der Grundlage von Phosphoinositid-Bindungsdomänen zugrunde (Kasten 1; Tabelle 1).”

GTPAsen? Hm… Helft mir mal kurz: Was war eines der Hauptaugenmerke, denen ich mich recht früh widmete? Ach ja! Richtig! Das RAS-Protein, K-RAS im speziellen.
Siehe mein Substack über mögliche Krebsrisiken.

Dazu gleich mehr. Doch zuerst noch ein wenig aus diesem großartigen Review:

”PtdIns wird im ER synthetisiert [Endoplasmatisches Retikulum] und wird über Lipidaustauschproteine an MCSs und durch vesikuläre oder tubuläre Träger, die sich entlang des sekretorischen Weges und innerhalb des endolysosomalen Systems bewegen, an andere Organellen abgegeben. Durch reversible Phosphorylierung und Dephosphorylierung des Inositolrings durch Phosphoinositid-Kinasen und -Phosphatasen entstehen sieben verschiedene Phosphoinositid-Spezies. Diese Spezies können je nach enzymatischer Aktivität ineinander umgewandelt werden, was zu einem Stoffwechselnetz mit vielfältigen gegenseitigen Abhängigkeiten führt (Abb. 1b). Phosphoinositide machen weniger als 1 % des gesamten Phospholipidpools der Zellen aus, weisen jedoch aufgrund der engen räumlichen und zeitlichen Kontrolle ihrer Synthese und ihres Umsatzes eine stark lokalisierte subzelluläre Verteilung auf (Abb. 1c). Dies wirkt sich direkt auf die Lokalisierung und Aktivität von zytosolischen Effektorproteinen und membranintegralen Proteinen wie Rezeptoren, Ionenkanälen oder Transportern aus, die mit diesen Lipiden interagieren und eine physiologische Reaktion auslösen (z. B. eine Signalkaskade oder die Bildung eines Proteinkomplexes, der die Membranen umgestalten kann). Phosphoinositid-bindende Domänen von Effektorproteinen binden im Allgemeinen mit geringer Affinität an ihr Ziellipid. Daher sind für die Funktion von Effektorproteinen eine multivalente gleichzeitige Erkennung zusätzlicher Faktoren, einschließlich kleiner GTP-bindender Proteine und anderer Lipide, sowie Aviditätseffekte erforderlich, die durch das Vorhandensein mehrerer Lipidbindungsstellen oder -domänen auf Effektorproteinen verursacht werden.”
Gehen wir mal die beiden hervorgehobenen Sätze durch:
“Dies wirkt sich direkt auf die Lokalisierung und Aktivität von zytosolischen Effektorproteinen und membranintegralen Proteinen wie Rezeptoren, Ionenkanälen oder Transportern aus, die mit diesen Lipiden interagieren und eine physiologische Reaktion auslösen (z. B. eine Signalkaskade oder die Bildung eines Proteinkomplexes, der die Membranen umgestalten kann).”
Mit einfachen Worten: Die Endozytose wird sich direkt auf sämtliche Proteinkanäle und membrangebundenen Rezeptoren auswirken und diese in ihrer Funktion und Lokalisation verändern (alterieren). Rezeptoren, wie die Toll Like Rezeptoren 1,2,4,5,6,10? Rezeptoren wie den vaskulär-endothelialen Faktor Rezeptor (VEGFR)? Rezeptoren wie den epidermalen Wachstumsfaktor Rezeptor (EGFR), den Transforming Growth Factor Rezeptor TGFR? Hm…. Wofür steht RAS noch mal?
http://www.eurekaselect.com/135441/article
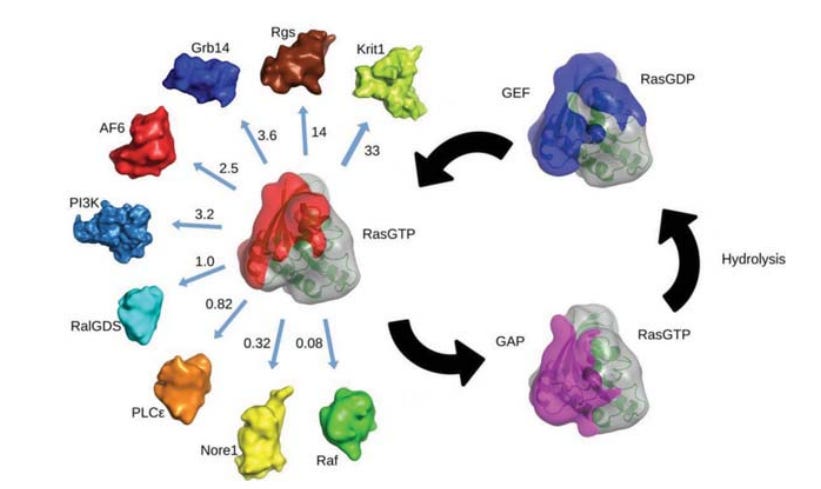
“RAS ist ein molekularer Schalter, der durch Interaktionen mit vielen Effektorproteinen eine große Anzahl von Signalwegen reguliert. Die meisten RAS/Effektor-Komplexe sind kurzlebig und weisen eine schnelle Assoziations- und Dissoziationsrate sowie KDs…” (Dissoziationskonstanten) “…von 10 hoch (-8) bis 10 hoch (-5) M auf, was mit der Signalfunktion dieser Wechselwirkungen in der Zelle vereinbar ist. Die RAS-Effektoren weisen eine geringe Sequenzhomologie auf, enthalten aber alle eine RAS-Bindungsdomäne, die eine Ubiquitin-Faltung aufweist. Alle Effektoren binden an dasselbe Epitop auf RAS, indem sie ein intermolekulares Beta-Sheet bilden und eine Reihe von günstigen Wasserstoffbrücken und Salzbrücken über die Bindungsschnittstelle hinweg schaffen. Mehrere Hotspots sowohl auf RAS- als auch auf Effektormolekülen bilden einen allgemeinen Erkennungsmodus. RAS/Effektor-Wechselwirkungen treten nur auf, wenn sich RAS im aktiven, GTP-gebundenen Zustand befindet, und werden bei der GTP-Hydrolyse unterbrochen, was wahrscheinlich auf die erhöhte Flexibilität des RAS-Moleküls zurückzuführen ist. Jüngste NMR-Studien zeigen, dass RAS bei Vorhandensein mehrerer Bindungspartner bestimmte Effektoren gegenüber anderen bevorzugt. Die Hierarchie dieser Wechselwirkungen könnte bei onkogenen RAS-Mutanten verändert sein, wodurch das Netzwerk der nachgeschalteten Signalübertragung gestört wird. Die durch biophysikalische und strukturelle Untersuchungen von Effektoren, die mit RAS und seinen Mutanten interagieren, gewonnenen Erkenntnisse bilden die Grundlage für die Entwicklung von Medikamenten gegen RAS-assoziierte Krankheiten.”
Ach ja: Aktivierte GTPase, die zu Wechselwirkungen mit Effektorproteinen führt? Und welches Protein gehört dazu? Oh PI3K? Die Phosphoionositid3-Kinase. Huch. Und die spielt also für die Membranorganisation eine entscheidende Rolle? Also dürfen wir schon mal davon ausgehen, dass PI3K nach der Transfektion in Mitleidenschaft gezogen wurde. Diese Hypothese werde ich später noch ein wenig näher beleuchten mit dieser Ausgangsfeststellung, dass PI3K als Effektorprotein von den RAS (Ratsarcoma-Superfamilie) angesteuert wird. Und wie ihr anhand dieses großartigen GTPase via RAS Schematik erkennen könnt, werdet ihr nicht RAS aktivieren können, ohne zwingend downstream (Richtung Nukleus in einer Zelle) die nächste daran geschaltete Signalinteraktion mit RAF zu triggern. Darum nennen wir es eben allgemeinhin Signaltransduktionskaskade. In diesem Fall also RAS→RAF→MEK→ERK-MAPk→welche im Regelfall NF-κB (darauf komme ich noch an späterer Stelle zurück) aktiviert.
“Daher sind für die Funktion von Effektorproteinen eine multivalente gleichzeitige Erkennung zusätzlicher Faktoren, einschließlich kleiner GTP-bindender Proteine und anderer Lipide, sowie Aviditätseffekte erforderlich, die durch das Vorhandensein mehrerer Lipidbindungsstellen oder -domänen auf Effektorproteinen verursacht werden.”
Verstehe ich diesen Satz richtig?: Die GTPasen (RAS inklusive und vermutlich sogar bevorzugt) + Aviditätseffekte (Anziehungseffekte) sind notwendig, um das für die Membranorganisation notwendige Effektorprotein zu aktivieren? Und dieses besitzt zeitgleich zwingend Lipidbindungsstellen? Und wir kacheln eine Lipidbombe - bestehend aus zig synthetischen Phospholipiden, unnatürlichen kationischen / ioniserten Lipiden, Cholesterol und PEG‼ylierten‼(wichtiges Wort) Lipiden rein? Woran würden sich dann wohl die Effektorproteine, wie PI3K, binden? Und warum würde zwangsläufig dadurch bedeutend mehr RAS rekrutiert werden müssen?
Ok… gehen wir zusammen mal die generelle Thematik der PI3K und RAS→RAF→MEK→ERK→MAPk-‼KASKADEN‼ durch:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3112285/

“Die Signalwege Ras-ERK (extrazelluläre signalregulierte Kinase) und PI3K (Phosphatidylinositol-3-Kinase)-mTOR (mammalian [laut aktuellem Stand “mechanistic”] target of rapamycin) sind die wichtigsten Mechanismen der Zelle zur Steuerung des Überlebens, der Differenzierung, der Proliferation, des Stoffwechsels und der Motilität von Zellen als Reaktion auf extrazelluläre Reize. Die Komponenten dieser Signalwege gehörten zu den ersten, die entdeckt wurden, als Wissenschaftler in den 1980er Jahren begannen, Proto-Onkogene zu klonen und zelluläre Kinaseaktivitäten zu reinigen. Ursprünglich wurden Ras-ERK und PI3K-mTOR als lineare Signalwege modelliert, die durch unterschiedliche Stimuli aktiviert werden, doch schon frühe Experimente deuteten darauf hin, dass sie sich überschneiden könnten, um sich gegenseitig zu regulieren und nachgelagerte Funktionen mitzubestimmen. Das Ausmaß dieses Crosstalks und seine Bedeutung für die Krebstherapie werden nun deutlich.”

Für dieses Substack ist an diesem Schaubild folgende Erklärung wichtig:
”Aktivierte PI3K phosphoryliert PIP2, um membrangebundenes PIP3 zu erzeugen. Pleckstrin-Homologie (PH)-Domänen in AKT und PDK1 erkennen PIP3 und wandern zur Membran. PDK1 phosphoryliert die Aktivierungsschleife und mTORC2 phosphoryliert das hydrophobe Motiv von AKT, wodurch die Aktivierung von AKT und die Phosphorylierung von TSC2 gefördert wird. Diese TSC2-Phosphorylierung hemmt die TSC2-GAP-Aktivität. RHEB-GTP lokalisiert sich im Lysosom und aktiviert mTORC1 nach seiner Rekrutierung durch die Rag-GTPasen.”
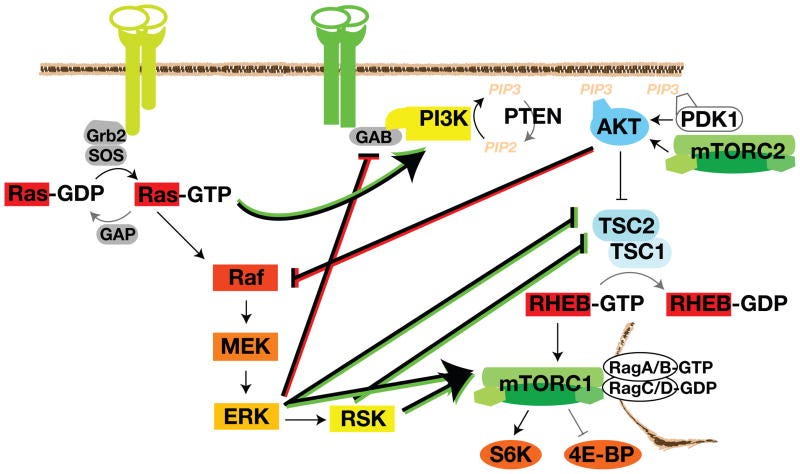
Und hier sieht man dann wunderschön, wie ERK direkt und über RSK mTORC1 ansteuert und das aktivierte GTP-gebundene RAS aktiviert PI3K. Während ERK GAB (ein GRB2-bindungsassoziiertes Bindemittel - GRB2 ist ein weiteres Signalprotein) inhibieren kann. Interessanterweise würde Akt unter bestimmten Bindungsbedingungen an mTORC2 gebunden RAF inhibieren. Na? - Gute Idee mal eben diese Signalproteine in den Kaskaden zu unnatürlichen Reaktionen zu forcieren? Und immer im Kopf behalten: Kaskaden sind wie Wasserfälle. Vllt. noch eine kleine Analogie: Habt ihr mal versucht mit 220 Sachen eine Vollbremsung auf der Autobahn zu machen? → Wenn dies die Reaktion der eigentlichen Kaskade wäre, dann stellt euch die Crosstalks als Seitenstreifen, Leitplanke Rück- und Gegenverkehr vor.
Einen weiteren Hinweis, dass der RAS-Signalweg eine absolut essentielle Rolle bei der Interaktion mit der Zellmembran spielt, wollen wir noch näher beleuchten:
Zur kurzen Erklärung, um den Text zu begreifen:
Eine SH2-Domäne ist eine Proteindomäne, die spezifische Proteininteraktionen vermittelt. Die Abkürzung SH2 steht für Src-homology 2 (abgeleitet von der Proteinkinase c-Src, in der die Domäne das erste Mal beschrieben wurde). Und damit sind wir dann schon bei Tyrosin angekommen.
https://ncbi.nlm.nih.gov/pmc/articles/PMC5776726/

Membranlipide und Zellsignalisierung
“Insbesondere kann die Zusammensetzung der Membranlipide regulieren, wie Proteine mit SH2-Domänen und Moleküle wie K-Ras ihre katalytischen Domänen dem Zytosol aussetzen und mit Effektoren und Second Messengern interagieren. Jüngste Berichte haben auch gezeigt, dass der Sättigungsgrad von Phospholipiden die Aktivierung bestimmter G-Protein-gekoppelter Rezeptoren sowie die dem Toll-like-Rezeptor 4 nachgeschaltete Signalübertragung verringern kann, was sich auf die NFkB-Aktivierung und Entzündung auswirkt. Es wurde berichtet, dass der Gehalt an spezifischen Gangliosiden in der Membran Integrine auf zellautonome Weise aktiviert und die Migration von Tumorzellen beeinflusst. Darüber hinaus konnte durch die hochauflösende Untersuchung der Assoziation von Cholesterol mit dem Smoothened-Rezeptor dessen Beteiligung an der Sonic-Hedgehog-Signalgebung geklärt werden. Dies sind einige der wichtigsten Fortschritte, die unser Verständnis der vielseitigen Beiträge von Membranlipiden zur Signaltransduktion weiter vorangetrieben haben.”
Ich denke, diesen Passus muss ich nicht noch groß übersetzen: Zusammenfassend heißt dies also, dass auch Cholesterol(auch falsch bezeichnet als Cholesterin) und Phospholipide (auch in der Lipidbombe von BNT162b2 enthalten) die intrazellulären Signalkaskaden regulieren und ansteuern können und zudem die Ladungssättigung der jeweiligen Lipide, die auf die Membran treffen Einfluss auf die weitere Transduktion haben wird.

“Darüber hinaus ist bekannt, dass Membranlipide bei der Zellsignalgebung als sekundäre Botenstoffe fungieren. Viele G-Protein-gekoppelte Rezeptoren aktivieren den Phosphatidylinositol-Signalweg, wobei Phospholipase C Phosphatidylinositol-4,5-Biphosphat (PIP2) in zwei sekundäre Botenstoffe hydrolysiert: Inositol-1,4,5-Triphosphat (IP3) und Diacylglycerol (DAG). DAG wiederum aktiviert die Proteinkinase C, und IP3 erhöht den intrazellulären Kalziumspiegel, was eine Vielzahl von Zellreaktionen fördert, darunter Transkription, Zellwachstum und Immunreaktionen. Interessanterweise hat sich gezeigt, dass ein anderes Membranlipid, Sphingosin, die Proteinkinase C moduliert.
In jüngerer Zeit hat sich gezeigt, dass Membranlipide die Signalübertragung durch integrale Membranrezeptoren entweder durch direkte oder indirekte stöchiometrische Wechselwirkungen verändern. Untersuchungen der letzten fünf Jahre haben gezeigt, dass Lipide eine wichtige Rolle bei der Regulierung von Membranproteinrezeptoren während der Zellsignalisierung spielen.”
stöchiometrische Wechselwirkungen: Massenverhältnisse in Wechselwirkung
Das Ganze ist sehr plausibel erklärbar, wie ich gleich noch zeigen werde: Wenn der RAS-RAF-MEK-ERK-MAPk-Signalweg und die anderen MAPks für cyclinabhängige Kinasen, Transkriptionsfaktoren, wie E2F und NF-κB, stehen und unter anderem Proliferation, Zellüberleben und Zellteilung mit regulieren, dann müssen diese Kaskaden direkte membranregulierende Funktionen auch übernehmen, indem sie Signale während der Endozytose zum Nukleus schicken. Wie schon erwähnt, reden wir von Kaskaden: Da unsere Zellen keine Smartphones besitzen, um mal eben dem Nukleus bescheid zu sagen, “schick mir mal, die und die mRNA zur Translation und Proteinbildung” müssen solch komplexe Signalwege runter gehen und punktgenau das zu bildende Produkt über aktivierte Transkriptionsfaktoren, wie NF-κB ansteuern. Und dies geschieht eben durch unglaublich komplexe ladungsabhängige Prozesse, die dies ermöglichen. Damit fährt dann beispielsweise - grob vereinfacht - die Expression von Gen XYZ rauf oder runter, in dem das Zielgen mehr/weniger/ gar nichts mehr expressiert. (Natürlich spielen noch mehr Komponenten, wie miRNA, Histonregulierung, etc mit rein).
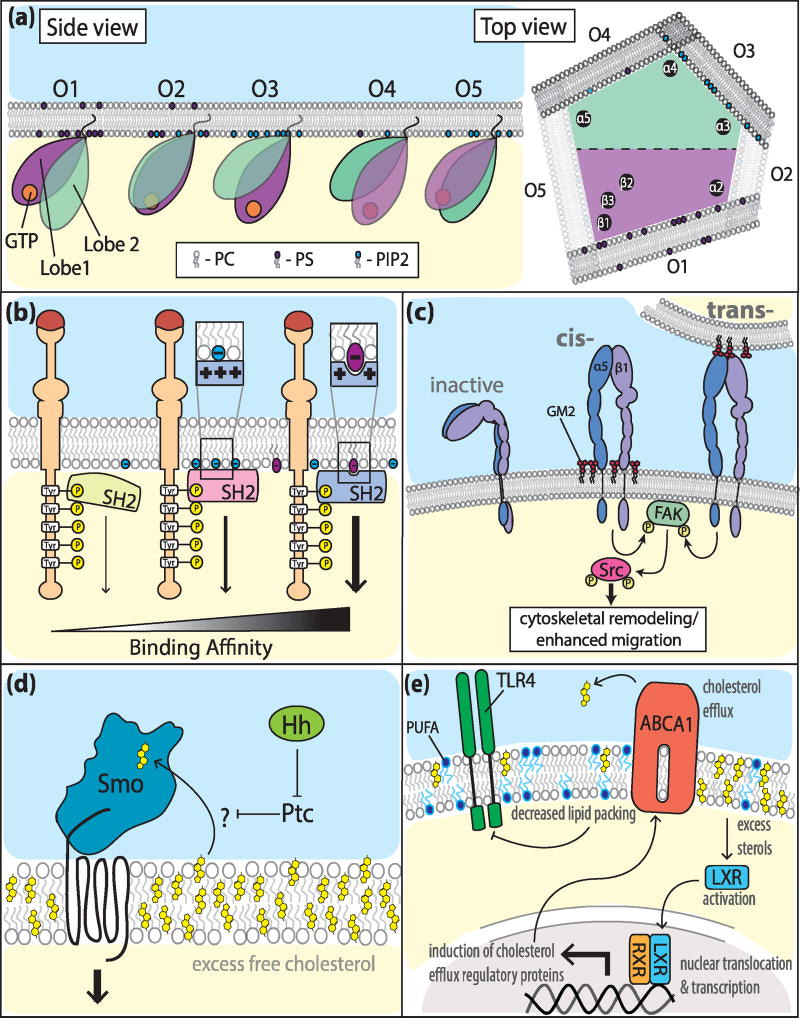
“Darüber hinaus können Lipidsubtypen vielseitige Verankerungsplattformen bieten, die Protein-Protein-Wechselwirkungen und die nachgeschaltete Signalübertragung modulieren. Eines der bekanntesten Module für Protein-Protein-Wechselwirkungen in der Zellsignalgebung sind die Src-Homologie-Domänen (SH). Vor allem die SH2-Domänen sind echte Bindungsmodule, die für die nachgeschaltete Signalübertragung in mehreren Phosphotyrosin-(pY)-Rezeptorwegen von wesentlicher Bedeutung sind. Vor zwei Jahrzehnten wurde berichtet, dass SH2-Domänen auf eine Weise an Lipide binden, die ihre Aktivität entweder hemmt oder fördert. Diese Erkenntnisse blieben jedoch umstritten, da es ihnen an mechanistischen Details mangelte. In einer aktuellen Studie nahmen Park und Kollegen die Herausforderung an und untersuchten systematisch die Rolle von Lipiden bei der Regulierung von SH2-Domänen-vermittelten Protein-Protein-Interaktionen und nachgeschalteten Signalereignissen. Sie lieferten stichhaltige Beweise dafür, dass die meisten SH2-Domänen mit hoher Affinität an PM (Plasma Membran)-Lipide binden, und zwar über ihre so genannten "alternate cationic patches" (ACPs), bei denen es sich um Domänen handelt, die an hydrophobe und/oder aromatische Reste angrenzen, die an membranbindende Proteinlipidbindungsstellen erinnern (Abbildung 1b). Die Studie zeigte, dass SH2-Module in der Tat eine doppelte Spezifität mit Lipid- und Proteinbindungsdomänen aufweisen. Auf diese Weise diktieren die relativen Positionen der ACPs und der pY-Tasche die Proteinausrichtung der SH2-Domäne und begünstigen flexible, lipidvermittelte Regulationsmechanismen für pY-Signalergebnisse auf der Grundlage der Lipidbindungsaffinität (Abbildung 1b). Die Autoren fanden außerdem heraus, dass die Morphologie der lipidbindenden ACPs es SH2-haltigen Proteinen ermöglicht, unterschiedlich mit der PM (plasma membrane) zu interagieren; rillenförmige ACPs binden an Lipidkopfgruppen, während flache ACPs unspezifisch mit anionischen Lipiden interagieren (Abbildung 1b). Zusammenfassend lässt sich sagen, dass diese unterschiedlichen Assoziationen die Beweglichkeit der SH2-haltigen Proteine und ihre Interaktion mit verschiedenen nachgeschalteten Effektoren beeinflussen.“
Grob und einfach übersetzt: Jede Form von Lipid, welches auf die Membran stößt, wird auch zwingend Signalkaskaden (insbesondere die, deren Signalproteine eine SH2-Bindungsdomäne haben) direkt oder indirekt an die Membran locken und in ihrer Aktivität positiv oder negativ ansteuern. Dies begründet sich damit, dass die Rezeptoren in ihrer Funktion angesteuert werden.
Etwas früher hielten die Autoren dieses brillanten Reviews fest, bei dem ich offen gestanden eine Gänsehaut und tiefste Ehrfurcht vor eukaryotischen Zellen empfinde:

“Mit Hilfe von Molekulardynamiksimulationen, die alle Atome berücksichtigen, fanden Li und Buck heraus, dass die Assoziation von K-Ras mit verschiedenen Phospholipiden seine Orientierung und damit seine Funktion moduliert. Die Autoren zeigten fünf mögliche zytosolische Topologien von Ras, die von der Art der anionischen Lipide an der Membran abhängen, was zu Veränderungen bei der Exposition der katalytischen Domäne und damit ihrer Fähigkeit zur Signalgebung führt. Interessanterweise scheint die Affinität von K-Ras zu Membranlipiden auch nukleotidabhängig zu sein.”
Wie sicher und wirksam ist also eine synthetische Lipidbombe, die jede Membran durchschneidet und penetriert und noch dazu kationische Lipide (+ geladen) (das Gegenstück zu anionischen) enthält?
Bevor ich darauf nun noch eingehen werde, und noch vorher die Affinität von RAS zeigen werde, an PIP2 zu binden, sollte es mutieren, muss ich ein kleines Kommunikationsproblem eingestehen, auf welches mich Geoffrey Norman Pain aufmerksam machte, um nicht Chaos auszulösen.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3546036/
Ich kann und will bestimmt nicht exkludieren, dass auch die BNt162b2-Lipidbombe endotoxische Effekte ‼nach‼ der Transfektion auslöst. Doch diese wären/ ?sind? zeitverzögert, nachdem die LNPs im Zytosol sind. Was wiederum absoluten Sinn ergibt, da - im Gegensatz zu Lipid A - dem Hauptauslöser der Endotoxischen Reaktion - diese Fettkugelbomben eben NICHT direkt den TLR4 ansteuern, sondern die Plasmamembran penetrieren werden (siehe weiter unten). Es wird also insofern vernachlässigbar für meine Problemstellung, da die Transfektion bittere Realität sein wird, noch bevor das Lipid in der Zelle verarbeitet wird, wie auch der Einfluss der 4 Lipide dieser Clusterbombe (dazu später noch ein wenig mehr via FOIA FDA und Distributionsstudien) auf die Membran downstream die Signalwege zum Nukleus affektieren wird. Die Problematik, der ich mich hier also widme, ist nicht die Frage, was NACH der Transfektion im Zytosol passiert während dem endosomalen Escape und der Translation, sondern zu fragen in welcher Art im Detail die Signalkaskaden affektiert sein könnten/ vermutlich sind und was mögliche Konsequenzen auf Genexpressionen sind. Eben jene Prozesse, welche schon während dem Eintritt ins Zytosol falsch getriggert wurden und bereits die gesamte Zellkommunikation durchwirbeln. Und dabei ist dann eben der Blick auf die Genexpression bedeutend präziser, als alleine auf Zytokine zu schauen. → Und dahingehend, kann ich nur mit mir bekannten Daten argumentieren.
Und um dies wirklich zu verstehen im Sinne, was geschah also nach Kontakt mit den LNPs, braucht es diese ellenlange Einleitung und Vorbereitung.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6169983/

Ras und die Plasmamembran: Eine komplizierte Beziehung
“Der Hauptort der Ras-Signaltransduktion ist die Plasmamembran (PM). An der PM teilen sich die ubiquitär exprimierten Ras-Isoformen, H-, N- und K-Ras, räumlich in nicht überlappende, nanometergroße Domänen auf, die als Nanocluster bezeichnet werden, mit einer weiteren lateralen Aufteilung in nicht überlappende Guanosintriphosphat (GTP)-gebundene und Guanosindiphosphat (GDP)-gebundene Nanocluster. Die Nanocluster von Ras haben unterschiedliche Lipidzusammensetzungen als Ergebnis der Lipidsortierungsspezifität, die in jedem carboxyterminalen Membrananker von Ras kodiert ist. Die Rolle der G-Domäne bei der Regulierung der Anker-Membran-Interaktionen wird immer deutlicher. ”
Hier wird noch mal im Detail beschrieben, wo die Isoformen von RAS zu finden sind, wie sie strukturell organisiert sind und welche Bindungseigenschaften sie haben.
“Die Rolle der G-Domäne bei der Regulierung der Anker-Membran-Interaktionen wird immer deutlicher. Die G-Domänen von Ras verändern bei einem Guanin-Nukleotidwechsel ihre Konformation erheblich, was zu unterschiedlichen direkten Kontakten zwischen der G-Domäne und einer Reorganisation des Membranankers führt. Ras G-Domänen enthalten auch schwache Dimerschnittstellen, was zur Homodimerisierung führt, die ein obligatorischer Schritt der Nanoklusterbildung ist. Die Modulation der Bildung von Ras-Dimeren, der Lipidzusammensetzung der PM oder der lateralen Dynamik wichtiger PM-Phospholipide stellen neuartige Mechanismen dar, mit denen das Ausmaß des Ras-Nanoclustering reguliert werden kann, um die Verstärkung der Ras-Signalkreisläufe zu steuern.”
Zusammenfassend lässt sich also sagen, dass die Membranphospholipide die RASproteine direkt in ihrer Signalstärke und Kreisläufen mit steuern.
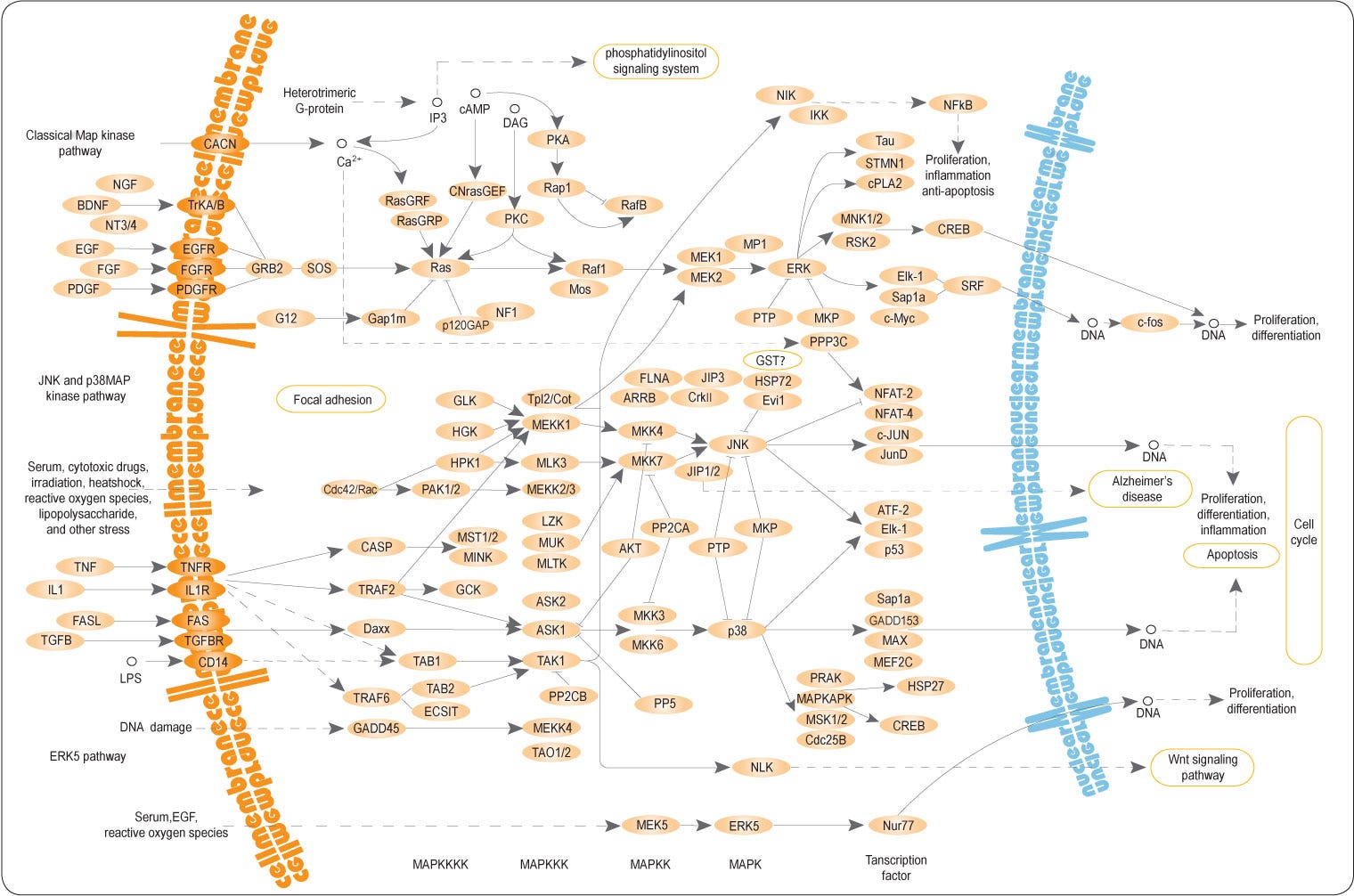
Und jetzt einfach mal diese wunderschöne Schematik der MAPkkk/MAPkk/MAPk downstream auf sich wirken lassen. Wenn ihr euch bewusst macht, dass dies eine einzige große Signalkaskadenfamilie ist und es noch unzählige andere gibt, wie beispielsweise den oben erwähnten Sonic-Hedgehog-Signalweg (SHH) und sie in jedem Prozess, der in eurer Zelle passiert zeit-und raumabhängige Interaktionen vollziehen und es bei allen mit den Rezeptoren oder aber, wie am Beispiel der RAS-RAF-MEK-ERK-Kaskade gezeigt, der Endozytose losgeht, hoffe ich, dass auch euch der Atem stockt. Wie wahrscheinlich wäre es also, etwas, wie die LNPs, ohne fatalste Konsequenzen für diese Zelle in ihren Basisfunktionen, einzuschleusen?
Siehe dieses Fallbeispiel für den SHH, was so schief gehen kann:
https://www.spandidos-publications.com/10.3892/ijmm.2021.4939

Wie man anhand dieses Beispieles erkennen kann, spielen auch hier Crosstalks zur MAPkkk/kk/k via PKA und ERK1/2 beispielsweise mit rein.
Habt ihr bis hier her aufgepasst und noch folgenden Satz “Aktivierte PI3K phosphoryliert PIP2, um membrangebundenes PIP3 zu erzeugen.” im Gedächtnis?:
Ein kleiner Funfact dazu ist nämlich das Faktum, dass, sollte K-RAS mutieren, es auch an PIP2 binden kann.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6497929/
Bindung der G-Domäne von K-Ras an das Signallipid Phosphatidylinositol(4,5)-phosphat (PIP2): Membranassoziation, Proteinorientierung und Funktion

“Wir entdeckten, dass die β2- und β3-Stränge sowie die Helices 4 und 5 der GTPase-G-Domäne an PIP2 binden, und identifizierten die spezifischen Rückstände in diesen Strukturelementen, die bei diesen Interaktionen verwendet werden und wahrscheinlich in zwei K-Ras4B-Orientierungszuständen relativ zur Membran auftreten. Wichtig ist, dass wir herausgefunden haben, dass einige dieser Reste, die als onkogen bekannt sind, wenn sie mutiert sind (D47K, D92N, K104M und D126N), für die K-Ras-vermittelte Transformation von Fibroblastenzellen entscheidend sind, aber die basale und unterstützte Nukleotidhydrolyse und den Austausch nicht wesentlich beeinflussen. Außerdem hob die K104M-Substitution die Lokalisierung von K-Ras an der Plasmamembran auf. Die Ergebnisse deuten darauf hin, dass spezifische G-Domänen-Reste die Funktion von Ras entscheidend regulieren können, indem sie Interaktionen mit membranassoziierten PIP2-Lipiden vermitteln. Diese Erkenntnisse können in Zukunft die Entwicklung therapeutischer Reagenzien beeinflussen, die auf die Aktivität von Ras abzielen.”
Weiter heißt es:
"Es ist daher wahrscheinlich, dass die Lokalisierung, wenn nicht sogar die Aktivität der Ras-GTPasen auch durch Veränderungen der PIP2-Konzentration in der Nähe beeinflusst wird und dass PIP2 selbst durch bestimmte Proteine lokal geclustert werden kann. Darüber hinaus ist PI3K ein wichtiger Effektor von Ras, der, wenn er von der GTPase aktiviert wird, PIP3 aus PIP2 erzeugt, während PLCβ und -γ von Ras reguliert werden, um PIP2 abzubauen. Möglicherweise sind PIP2 und Ras an positiven oder negativen regulatorischen Rückkopplungsschleifen beteiligt. Es ist bekannt, dass PIP2 eine Rolle bei der Lokalisierung von K-Ras in Zellen spielt, aber der Mechanismus ist umstritten und bleibt unklar (z. B. Ref. 33). Auf der Proteinebene haben wir die Möglichkeit, die molekularen (strukturellen/dynamischen) Details der Interaktionen zu verstehen, und insbesondere im Zusammenhang mit der HVR-Isoform- und Kompartiment-spezifischen Ras-Signalisierung beginnen wir gerade erst, diese Regeln zu entschlüsseln.”
Witzig: 2019 stellt man nach wie vor fest - und die Forschung läuft ja nun schon ein “paar Jährchen” - ,dass man gerade mal anfängt die Signaltranduktionskaskade über RAS zu begreifen. Selbiges gilt für jede andere Kaskade, in die ihr euch einlesen könntet, ebenso. Sie alle befinden sich in einem Netzwerk aus Crosstalks für Check and Ballance, damit es eben nicht zu chaotischen Genexpressionen kommt.
Wer den Komplex der Phospholipide auch noch aus biochemischer Reaktionsperspektive verstehen will, wo ich mich dann definitiv überfordert fühle: Hier ein Lernvideo.
Dieses großartige Video hilft ungemein.
Puh. Soweit so gut.
Wer noch mehr Lektüre darüber lesen möchte, wie die Plasmamembran die ganzen Rezeptoren organisiert, hier noch ein paar nette Papers, die euch ein besseres Bild für das Basisprinzip vermitteln können:
https://pubmed.ncbi.nlm.nih.gov/34242725/

Ein kleiner Funfact noch am Rande zu dem EGFR: Was aktivierte der gleich wieder?:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8563293/

“Die EGFR-RAS-MAPK-Signalübertragung ist auf die Plasmamembran und die damit verbundenen Endorecycling-Ausstülpungen beschränkt
Ras-GTPasen sind membranassoziierte Schlüsselelemente des EGFR-MAPK-Signalweges. Surve et al. zeigen, dass endogenes KRAS und NRAS vorwiegend an der Plasmamembran und ihren röhrenförmigen Ausstülpungen lokalisiert sind und dass ein kleiner Pool von Oberflächen-EGFRs die Signalübertragung entlang des RAS-MAPK-Signalwegs aufrechterhält, während internalisierte EGFRs nicht wesentlich zur MAPK-Aktivität beitragen.”
Wie ich schon weiter oben anmerkte: Raum-Zeit-abhängige Interaktionen.
https://pubmed.ncbi.nlm.nih.gov/26536269/

https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7830256/

(Lipid Rafts sind Mikrodomänen in der Membran)
Warum also RAS? Als Reminder aus meinem oben verlinkten Substack zu KRAS und der MAPk:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9029844/


Schauen wir uns nochmal dieses gruselige Werk an, was euch diese Expressionsalterationen der Gene als Erfolgsgeschichte verkaufen möchte:
Sechs Wochen nach der Pikse war KRAS - ein normalerweise wirklich kurzlebiges Signalprotein noch überexpressiert und aktiviert. Ja, wo aktiviert draufsteht, ist aktiviert drin, liebe “die Wissenschaft”™: Da dürft ihr auch gerne damit prahlen, schon publiziert zu haben. (Vielleicht scheitert es ja am Lesen der Argumente, die nicht den eigenen Annahmen und Publikationen entsprächen?)
Es gibt natürlich besonders “Schlaue”™ - ausgerechnet unter den Akademikern, die - vermutlich aufgrund von Forschungsgeldabhängigkeit, Arroganz und sonstigen Interessenskonflikten - euch erzählen möchten, dass eine Überexpressierung eines Signalproteins nichts weiter sei. Warum liest man dann jedoch überall, dass Signalkaskaden, wie die MAPkkk/kk/k eng reguliert sind und bei einer Fehlschaltung in zahllose Krankheiten, inklusive Krebs, Autoimmunerkrankungen und Creutzfeldt-Jakob involviert wären? Zur Veranschaulichung hier noch ein Auszug aus einer Konversation mit Dirk Fasshauer. (Akademiker aus dem Lehrbuch?)
https://twitter.com/NarfGb/status/1754920861197357557

Aber gerne doch, Dirk:
https://linkinghub.elsevier.com/retrieve/pii/0016-5085(87)90264-2

https://nature.com/articles/bjc2012477

https://oncotarget.com/article/19798/text/

https://cancerci.biomedcentral.com/articles/10.1186/s12935-015-0209-x

https://cell.com/cell-reports/pdfExtended/S2211-1247(15)00656-7


https://pubmed.ncbi.nlm.nih.gov/35120522

https://spandidos-publications.com/10.3892/ijmm.2017.2903

Oh, oh, armer Dirk. Ich bin unqualifiziert, weil ich nie publiziert habe, mich der Realität und komplexen Problemen zu stellen? Nun ja. Zu verbuchen unter “Follow the science”™-Impfpropaganda.
Wie also gerade gezeigt, war KRAS aktiviert und überexpressiert und es wurde mit der 3. Spritze von BioNtech sogar noch schlimmer in der Publikation von Knabl et al., welche schon ende 2021 als Preprint erschien.
Nun ist diese Studie jedoch nur bedingt brauchbar für mein Ausgangsstatement, dass die ersten Alterationen mit der Transfektion geschehen würden: Zum einen aufgrund der modRNA, welche auch noch mal im Zytosol richtig reinfeuern wird nach endosomalem Escape. Zum anderen aufgrund möglicher endotoxischer Reaktionen. Noch dazu haben wir das Impfspike als extremes Pathogen. Nichtsdestotrotz ist diese Publikation ein guter Indikator, dass etwas Furchtbares mit den Signalkaskaden der MAPkkk/kk/k, den JAK/STATs und mTOR geschehen sein muss.
Schauen wir uns also noch ein wenig Lektüre aus der Vergangenheit an, bevor ich auf die Penetration der Membran näher eingehen werde:
Ein kleiner Funfact noch: Lipide sind auch direkt bei der RNA-Regulierung beteiligt.
https://www.pnas.org/doi/full/10.1073/pnas.2119235119

“Alternativ könnten direkte RNA-Lipid-Wechselwirkungen die RNA an Membranoberflächen lokalisieren, ihre lokale Konzentration erhöhen und die Dimensionalität für intermolekulare Wechselwirkungen verringern. Schließlich bringt die Lokalisierung an einer Lipidoberfläche die RNA in eine physikalisch-chemisch einzigartige Mikroumgebung mit starken Gradienten der Hydrophobizität, der elektrischen Permittivität und der Wasseraktivität. Durch diese Effekte könnten RNA-Lipid-Wechselwirkungen einen leistungsfähigen Mechanismus zur Modulation der RNA-Aktivität darstellen.
Die erste funktionelle RNA-Lipid-Wechselwirkung wurde vor mehr als 40 Jahren beschrieben, und spätere Forschungen haben verschiedene Faktoren aufgedeckt, die die Bindung von Nukleinsäuren an Lipide erleichtern. In jüngerer Zeit wurden durch systematische Entwicklung von Liganden durch exponentielle Anreicherung (SELEX) spezifische RNA-Sequenzen mit Affinität für Flüssigkeitsmembranen, die aus Phospholipiden und Cholesterin bestehen, erzeugt.”
Summa summarum: RNA haben auch eine Bindungsaffinität zu Phospholipiden und Cholesterin. Fragt sich gerade noch jemand, was für ein Frankensteinspike ihr nach der Transfektion translatieren (übersetzen) würdet?
Doch das ist heute nicht das Thema. Zurück zum Signalkaskadenproblem:
Dass meine Annahme von alterierten Signalkaskaden mehr als eine bloße Theorie ist, wurde auch schon 2012 bestätigt - und seitdem gekonnt ignoriert- ,als man anfing mit kationischen Lipiden rumzuexperimentieren:
https://www.sciencedirect.com/science/article/abs/pii/S0169409X12001901




“Mitogen-aktivierte Proteinkinase-Kaskaden (MAP-Kinasen) bilden ein Netzwerk aus mehreren Serin/Threonin-spezifischen Proteinkinasen, die verschiedene zelluläre Prozesse regulieren, darunter Genexpression, Mitose, Differenzierung, Proliferation, angeborene Immunität, Zellüberleben oder Apoptose.
Die Inkubation von Makrophagen oder dendritischen Zellen mit kationischen Lipiden führte zur Aktivierung der p38 MAPK, der c-jun N-terminalen Kinase (JNK) und der extrazellulär-signalregulierten Kinase (ERK 1/2). Die MAPK-Aktivierung durch kationische Liposomen hängt auch von der Erzeugung von ROS (reaktiver Sauerstoffstress) ab, da ROS-Inhibitoren (N-Acetylcystein, TEMPO, DPI und Ebselen) sowohl die durch SA- oder DOTAP-Liposomen ausgelöste ERK- als auch die p38-MAPK-Phosphorylierung hemmten.”
Auf das P38-Signalprotein der MAPk komme ich noch zurück. Wie bereits oben ausgeführt: Kinase heißt nicht umsonst Kinase. RAS→RAF→MEK→ERK. Und weiter heißt es:
“Einige kationische Liposomen induzieren die Sekretion einer größeren Anzahl von Zytokinen. DiC14-Amidin-Liposomen induzieren die Sekretion von IL-1β, IL-6, IL-12p40, Interferon-β (IFN-β), Interferon-γ-induzierbarem Protein 10 (IP-10) und Tumor-Nekrose-Faktor α (TNF-α) durch myeloische dendritische Zellen von Mensch und Maus. Diese Eigenschaft, die vom TollLike Receptor 4 (TLR4) abhängt (siehe Abschnitt 3.1), wird von anderen kationischen Lipiden wie Lipiden der TAP- und EPC-Familien und DDAB nicht geteilt. Dies deutet darauf hin, dass nur einige wenige kationische Lipide, ähnlich wie diC14-Amidin, die TLR4-spezifischen strukturellen Anforderungen erfüllen und TLR4-abhängige Signalwege aktivieren.”
Nun werden einige fragen: Warum wird der TLR4 dann aber dennoch alteriert werden und warum betone ich immer wieder, dass ALLE membrangebundenen Rezeptoren affektiert sein werden? Was die Autoren hier nicht berücksichtigten (vielleicht, weil die Erkenntnis fehlte, vielleicht, weil sie im Wahn über die Begeisterung an der Tech bestimmte Details ausblenden wollten?), da auch hier mal wieder Folgestudien und vor allem Langzeitfolgestudien fehlen:
https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0051243

“Die Aktivierung von TLR löst komplexe Signalkaskaden aus, die je nach TLR und Zelltyp zu einer Reihe von entzündungsfördernden Ereignissen führen. Die Signalübertragung über die MyD88-abhängigen und -unabhängigen Signalwege ist zwischen Säugetieren und Hühnern weitgehend konserviert, obwohl TLR4 bei Hühnern möglicherweise nicht über den MyD88-unabhängigen Signalweg signalisiert. In Makrophagen haben sich evolutionär konservierte Signaltransduktionswege als Vermittler von Entzündungsprozessen erwiesen, darunter durch Mitogen-aktivierte Proteinkinasen (MAPK) induzierte Effektormechanismen. Es gibt drei Hauptgruppen von MAPK: die durch extrazelluläre Signale regulierte Proteinkinase 1/2 (ERK), die p38-MAP-Kinasen (p38) und die c-Jun amino-terminale Kinase (JNK), die viele zelluläre Funktionen, darunter auch Entzündungen, unterschiedlich regulieren. Mehrere TLR induzieren den Phosphoinositid-3-Kinase (PI3K)-Akt-Stoffwechselweg, der die Immunantwort auf negative oder positive Weise regulieren kann. Die Glykogensynthase-Kinase 3 (GSK3) ist ein nachgeschaltetes Ziel des PI3K-Akt-Signalwegs und ist für die Regulierung der Zytokinproduktion nach TLR-Aktivierung verantwortlich. Weitere Signalmoleküle, die an TLR-vermittelten Reaktionen beteiligt sind, sind die Janus-Kinase (JAK)-Familie, das durch doppelsträngige RNA (dsRNA) aktivierte Serin/Threonin-Kinase R (PKR)-Protein und die Serin/Threonin-Kinase Proteinkinase A (PKA). Das Ergebnis der TLR-Aktivierung ist somit eine Folge der Wechselwirkungen zwischen mehreren Signalwegen, von denen viele bei der Feinabstimmung der Entzündungsreaktion zusammenwirken.”
Übrigens gelten diese Crosstalks der MAPk ebenfalls für die zytosolischen RIG-1-Like Rezeptoren, NOD-Like und viele weitere.
Soweit so schick. Ich denke damit hätten wir die Frage geklärt, wie wahrscheinlich es ist, dass, wenn etwas in eine Zelle via Endozytose oder - im Falle der Transfektion - endozytoseähnlichem Prozess gelangt, downstream Signalkaskaden aktiviert werden. Doch wie komme ich nun auf die Aussage, dass eine Genexpressionsanalyse weit mehr über die falsch getriggerten Signale nach der Transfektion aussagen wird, als ein reines Zytokinprofil nach wenigen Stunden?:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3457779/

“Der Mitogen-aktivierte Proteinkinase (MAPK)-Signalweg umfasst verschiedene Signalkaskaden, von denen die Ras-Raf-Mek-extrazelluläre signalregulierte Kinase 1 und 2 (ERK1/2) eine der am stärksten dysregulierten bei menschlichem Krebs ist. Dieser Signalweg reguliert zahlreiche kritische Zellfunktionen wie Proliferation, Wachstum und Seneszenz. Das Ras-Gen ist ein wichtiger Bestandteil der großen Familie der GTPasen. Die Ras-Gene sind transformierende Onkogene, die erstmals in den 1960er Jahren von Jennifer Harvey (Harvey-Ras [H-RAS]) und Werner Kirsten (Kirsten-Ras [K-Ras]) als murine Sarkomviren erkannt wurden. Die Assoziation von aktivierten und transformierenden RAS-Genen bei menschlichem Krebs wurde 1982 von mehreren Autoren gleichzeitig berichtet. Spätere Studien führten zur Identifizierung eines dritten menschlichen RAS-Gens, das in menschlichen Neuroblastomzellen als NRAS bezeichnet wird (Neuroblastoma-Ras [N-Ras]). Die drei menschlichen RAS-Gene kodieren für vier eng verwandte Proteine mit 188 bis 189 Aminosäuren, die als H-RAS, N-RAS und K-RAS (K-RAS4A und K-RAS4B) bezeichnet werden. Ras-Proteine fungieren als binäre molekulare Schalter, die intrazelluläre Signalwege steuern, die an grundlegenden zellulären Prozessen wie Zellpolarität, Proliferation, Differenzierung, Adhäsion, Migration und Apoptose beteiligt sind. Ras und mit Ras verwandte Proteine sind bei Krebserkrankungen häufig dysreguliert, und zwar durch aktivierende Mutationen der Ras-Isoformen oder ihrer Effektoren bei fast einem Drittel aller menschlichen Krebserkrankungen. Ras aktiviert mehrere Signalwege, darunter die RAF-MEK-ERK/MAPK-Kaskade, die Signale stromabwärts weiterleitet und zur Transkription von Genen führt, die an der Steuerung verschiedener zellulärer Mechanismen beteiligt sind. Die Mitglieder der Ras-Familie sind an der zytoplasmatischen Seite der Plasmamembran durch carboxylterminale Farnesylierung [posttranslationale Modifikation von Proteinen] verankert. Durch diese Lokalisierung befindet sich Ras in unmittelbarer Nähe von Adaptoren, dem an den Wachstumsfaktorrezeptor gebundenen Protein 2 (Grb2) und dem Nukleotidaustauschfaktor son of sevenless (SOS), um den Austausch des an Ras gebundenen Nukleotids Guanosindiphosphat (GDP) mit Guanosintriphosphat (GTP) im Zytosol zu mildern. Durch diesen Austausch wird die Konformation von Ras aktiviert, was seine Interaktion mit einer Reihe von nachgeschalteten Effektoren ermöglicht. Dementsprechend übermittelt Ras externe zelluläre Signale an den Zellkern, und seine veränderte Aktivierung führt zu unangemessenen zellulären Aktivitäten, einschließlich verstärktem Zellwachstum, Differenzierung und Überleben und schließlich zu Krebs. Der RAS-RAF-MEK-ERK-Signalweg wird durch mehrere bekannte Wachstumsfaktoren und Zytokine aktiviert, die über Rezeptortyrosinkinase-Signale und durch aktivierende Mutationen hauptsächlich in den RAS- und RAF-Genen wirken.”
Hoppla: Die haben wirklich gesagt, dass RAS auch bei SOS-Signalen mit Neigung angezogen zu werden, entdeckt wurde, richtig? Seht es mir nach, dass ich keinen Bock habe, noch die Caspase 1 bis 9 durchzuackern.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4161249/

Und auch noch wichtig ist dieses lustige Zitat:
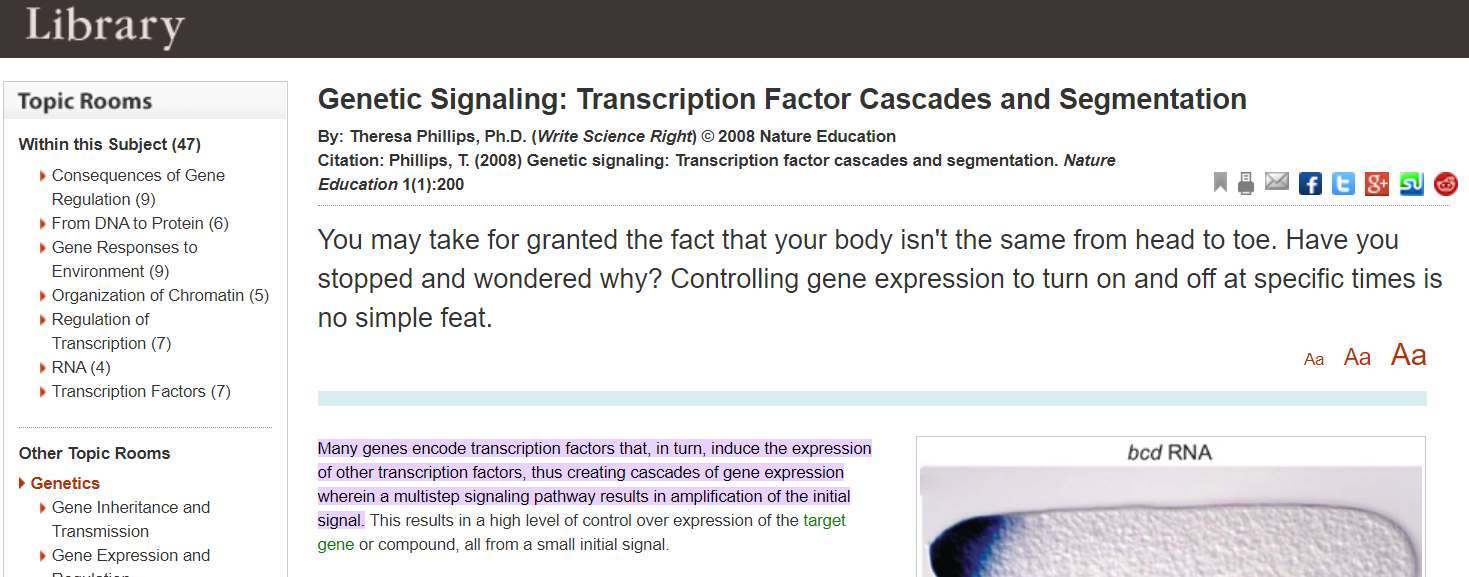
”Viele Gene kodieren für Transkriptionsfaktoren, die ihrerseits die Expression anderer Transkriptionsfaktoren induzieren, wodurch Kaskaden der Genexpression entstehen, bei denen ein mehrstufiger Signalweg zu einer Verstärkung des Ausgangssignals führt. Dies führt zu einem hohen Maß an Kontrolle über die Expression des Zielgens oder der Zielverbindung, und zwar ausgehend von einem kleinen Ursprungs/Initialsignals.”
Ich hab’ ne tolle Idee: Ignorieren wir doch einfach diese Erkenntnis, gewonnen aus über 100 Jahren Zellbiologie!
Puh: Das Gröbste ist geschafft! NF-κB (Oder auch liebevoll NF “trigger mich nicht, wenn du keinen Plan hast, wie ich funktioniere”-κB genannt)
Ich hätte noch viele weitere Papers einbauen können, die zeigen, wie Signaltransduktionswege durch Lipide beeinflusst werden. Doch ich denke, bis hier hin habe ich eine logische Kausalkette aufgezeigt und das Grundproblem dargestellt. Ein kleiner Funfact noch fix dazu:
Das PEI erklärt euch, mit seinen eigenen Worten, dass LNPs die Nachfolgetech von liposomalen Medikamenten wären.
https://www.pei.de/SharedDocs/Downloads/DE/newsroom/mitteilungen/201223-stellungnahme-empfehlung-allergiker.pdf?__blob=publicationFile&v=6

“Die Lipidnanopartikel ähneln den bereits seit vielen Jahren pharmazeutisch eingesetzten Liposomen, die als Träger für Arzneistoffe dienen. Einige der zugelassenen Liposomen/LNP haltigen Arzneimittel enthalten ebenfalls ein PEGyliertes Lipid (z.B. in Caelyx pegylated liposomal® oder Onpattro®).”
Aber liebes PEI: Dann sollte euch ja bestens bekannt sein, dass liposomale Medikamente nicht unbedingt eine Erfolgsgeschichte waren, wie ihr sie hier verkauft. Und ihr wollt höchstexperimentelle und immer noch nicht sonderlich gut verstandene Krebstherapien als Beispiel für “Transfektion ist toll” verkaufen. Nur ein einziges der gelisteten Medikamente kam bei einer wirklich lebensbedrohlichen Pilzinfektion zum Einsatz. Und bereits bei den Liposomen bestand das nicht gelöste Problem von alterierten Signalkaskaden nebst der Problematik mit der Biodistribution und Toxizität:
Guckste, du deine Aufgaben vernachlässigende, pharmabezahlte Schwurbelfabrik von “Die Experten”™:
https://patentimages.storage.googleapis.com/de/67/95/27ec2ea27a32d9/US10485884.pdf

https://pubmed.ncbi.nlm.nih.gov/36837644/

Und wer sich fragt, was Reprogrammieren heißt: Umfunktionalisierung. Ergo: anders geartete Basisfunktionen als Folge von nachhaltig anders getriggerter Genexpression.
Puh. Bevor ich nun in die Vollen gehe und wir das Ganze wasserdicht auf die BNT162b2- Transfektionsbrühe exemplarisch festnageln, fassen wir also bis hier hin noch mal das Gelernte zusammen:
Die Zellmembran und deren Lipid-Struktur ist maßgeblich an den Eingangssignalwegen und deren Regulierung sowie der Regulierung der membrangebundenen Rezeptoren und deren Signalgebung beteiligt. Signaltransduktion wiederum ist entscheidend involviert bei der Membranreorganisation nach Endozytose. Und jede Signalkaskade wird zwingend downstream weiter zu Transkriptionsfaktoren, wie NF-κB und der E2F-Familie gehen und damit die Genexpression ansteuern und feintunen. (Wir bleiben beim Beispiel RAS-RAF-MEK-ERK, um diese Aussage noch ein wenig zu untermauern.)
https://www.sciencedirect.com/science/article/pii/S0021925818527459

Die zeitliche Kontrolle der NF-κB-Aktivierung durch ERK reguliert die Interleukin-1β-induzierte Genexpression in unterschiedlicher Weise
“Die Hemmung von ERK hatte keinen Einfluss auf die Interleukin-1β-induzierte I-κBα-Phosphorylierung und den Abbau, dämpfte aber den I-κBβ-Abbau. Obwohl also die NF-κB-Aktivierung für die Interleukin-1β-Induktion jedes der untersuchten Proteine wesentlich war, wurde die Genexpression durch ERK und durch die Dauer der NF-κB-Aktivierung unterschiedlich reguliert. Diese Ergebnisse offenbaren eine neue funktionelle Rolle für ERK als wichtiger zeitlicher Regulator der NF-κB-Aktivierung und der NF-κB-abhängigen Genexpression.”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4189447/

https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2174281/

https://www.nature.com/articles/oncsis201681

Viele Variablen führen zu absolut engmaschig regulierten Prozessen:
https://www.mdpi.com/2075-1729/13/11/2150

Falls ihr euch die Mühe macht, diese Papers im Detail zu lesen und zu studieren, werdet ihr feststellen, dass jede Signalkaskade auch absolut zelltyp-, raum- und zeitabhängige, sowie interaktionsspezifische Einmaligkeiten zeigt.
Tusch, Paukenschlag und Trommelwirbel bitte!
https://www.britannica.com/science/cell-membrane

https://ncbi.nlm.nih.gov/pmc/articles/PMC8791091/

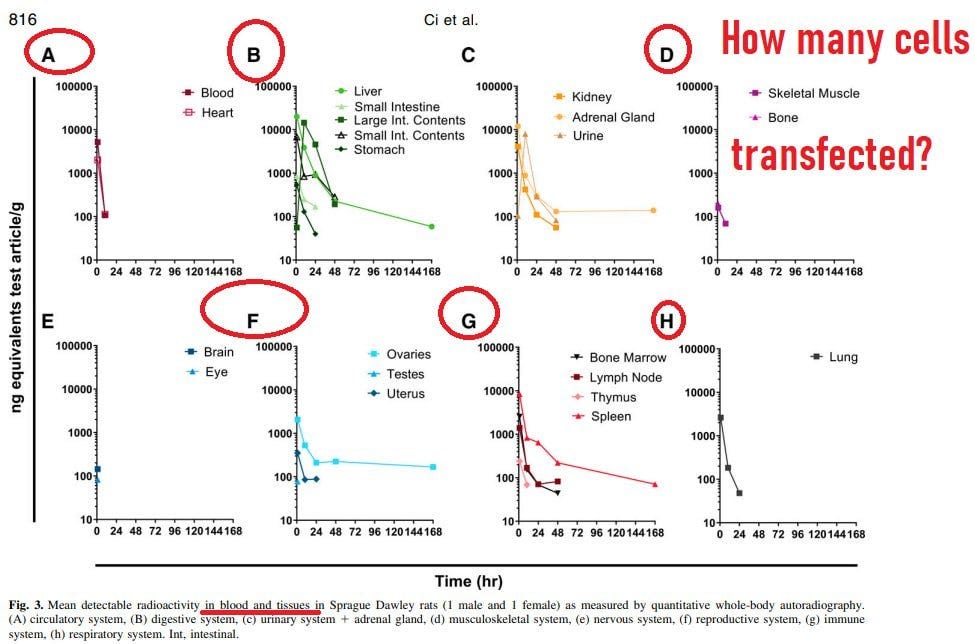
Hui ist das toll, wie es überall auch nach 48 Stunden noch aufflackert. Insbesondere im Hirn nimmt es sogar zu. Klasse! Bin ich froh, dass es im Muskel bleibt. Um die Genexpressionsalterationen zu begreifen, müssen wir auch verstehen, wohin die LNPs gehen. Dazu ackern wir uns noch durch ein paar Publikationen.
“Biodistribution und nicht-lineare Genexpression von mRNA-LNPs in Abhängigkeit von Verabreichungsweg und Partikelgröße
(…)
Einige intramuskulär injizierte LNP zirkulierten im Körper, was zu einer Anhäufung in Leber und Milz führte, insbesondere wenn die LNP relativ klein waren. Größere LNPs verbleiben eher an der Injektionsstelle. Im Vergleich zu anderen Organen und Geweben war die Expression von Transgenen in der Leber am stärksten ausgeprägt.
Schlussfolgerungen
Biomoleküle wie mRNAs, die in lokal injizierte LNPs eingekapselt sind, können über den Blutkreislauf in andere Organe und Gewebe gelangen. Die Genexpression wird durch die Biodistribution und Pharmakokinetik (PK) der LNP beeinflusst, die wiederum von der Partikelgröße und dem Injektionsweg abhängen. Da die Transfektionseffizienz in verschiedenen Organen variiert, sind die LNP-Exposition und die mRNA-Expression nicht linear korreliert.”

Gott sei Dank sind ja die Lipidformulierung von BNT162b2 und mod(e)RNA niemals nie nicht variierend in den Größen, was eine Shit happens-Bingo zur Folge hätte.
Und weiter heißt es:
”So wies die Leber die höchsten Transgenexpressionswerte auf, mehr als die Werte im Muskel, der eine höhere Verteilung von LNPs aufwies (Abb. (Abb.2).2). Sogar die Milz und die Lymphknoten wiesen höhere Transfektionseffizienzen auf als der Muskel, obwohl sie zu klein waren, um in den Biolumineszenzbildern des lebenden Tieres gesehen zu werden (Abb. (Abb.4A).4A). Nur die Luziferaseaktivität im Lymphknoten schien mit der Zeit stetig zuzunehmen. In anderen Geweben erreichte sie 6-10 Stunden nach der Injektion ihren Höhepunkt, danach nahm das Signal ab.”

Bitte beachtet, dass die Translationsaktivität (die Färbungen von Blau nach Rot) im Hirn zunahm und auch im Mäuseschwanz sah man mehr Aktivität. Und auch nach 72h war noch Aktivität beobachtbar, wenn auch abnehmend.
Ein wirklich bemerkenswerter Satz, den Ihr im Hinterkopf an späterer Stelle abrufbereit haben müsst:
“Die LNP-Exposition und der Gehalt an transgenen Proteinen waren nicht linear korreliert. Diese nicht lineare Beziehung zwischen der LNP-Exposition und dem Proteinexpressionsniveau variiert in verschiedenen Geweben und Organen. Darüber hinaus, wie in Abb. 6,6 gezeigt, selbst wenn sich mehr kleine Partikel in der Leber anreichern konnten als die mittleren Partikel, wurden die Luciferase-Aktivitäten in der Leber nicht stärker.”
Das heißt also, dass mehr LNPs die Organe erreichten, als Marker-RNAs in die Zelle eingeschleust wurden. Ich teile den Optimismus der Autoren jedoch nicht, darauf bauend Rückschlüsse auf die Transfektionsereignisse ziehen zu können. Denkt daran, dass wir Transfektion und endosomalen Escape klar differenzieren müssen und es mir in diesem Substack um die Transfektion und daran geschalteten Signaltransduktionen geht.
https://tga.gov.au/sites/default/files/foi-2389-06.pdf

Läuft doch? Äh… Also läuft durch und geht überall hin (sogar effizienter, als mein Kaffee. :-) )
https://dmd.aspetjournals.org/content/dmd/51/7/813.full.pdf?with-ds=yes
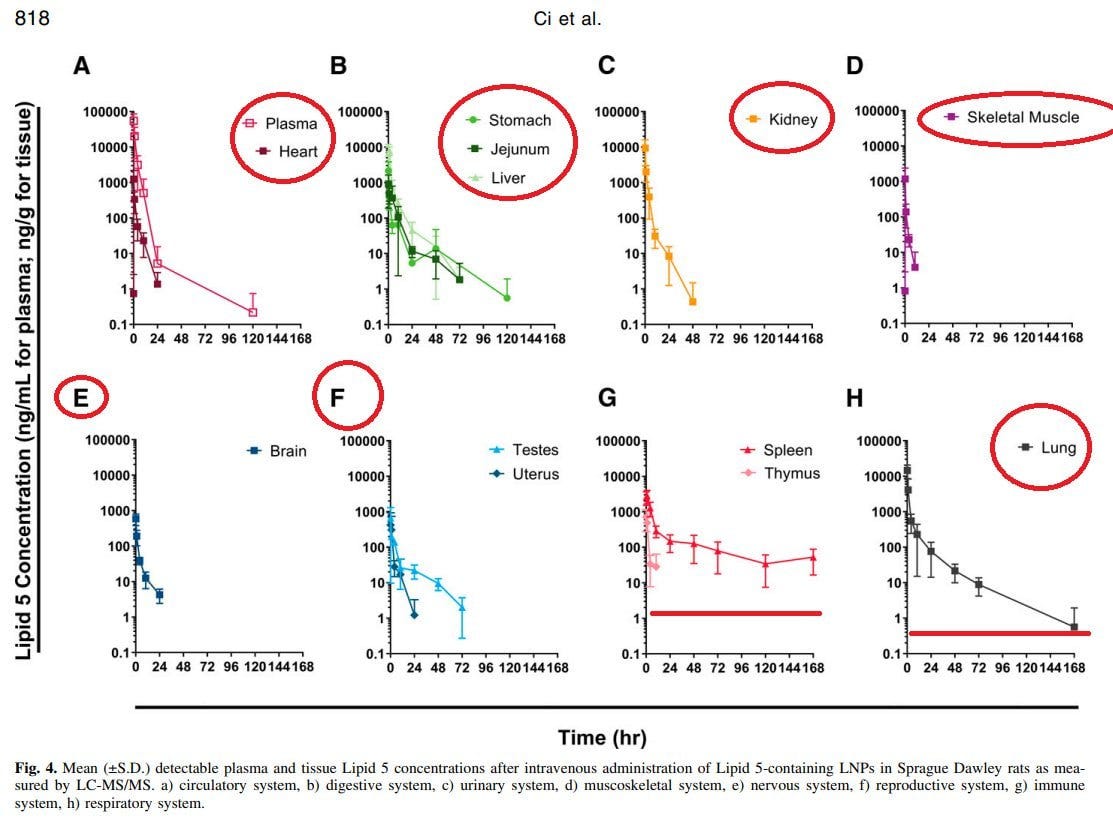
Biodistribution of Lipid 5, mRNA, and Its Translated Protein Following Intravenous Administration of mRNA-Encapsulated Lipid Nanoparticles in Rats
Und um das Ganze abzurunden, gibt es hier noch eine weitere Biodistributionsstudie mit anderen LNP-Formulierungen. Die Verteilung wird vermutlich ein wenig durch die Injektionsart abweichen. Nichtsdestotrotz ist dieses Paper eines der Besten zur Verteilung, die jemals publiziert wurden.
Und damit sind wir nun endlich auch beim allseits beliebten Event Transfektion angekommen. Es war ein langer Ritt bis hier hin und ich hoffe, dass ihr nicht zwischendrin “Kopf haut Tastatur” wegen der wahnsinnigen Komplexität gemacht habt.
Ich weiß nicht, wie es euch geht, aber wenn ich lese, dass etwas penetriert wird, löst das im Normalfall kein sonderlich gutes Gefühl bei mir aus?
https://www.sciencedirect.com/science/article/pii/S0009308423000166

Ionizable lipids penetrate phospholipid bilayers with high phase transition temperatures: perspectives from free energy calculations
Ich zitiere mal die wichtigsten Sätze aus diesem Chemiehöllentrip:
”Es wurde festgestellt, dass ALC-0315 am stärksten an die Membran bindet, während DLin-MC3-DMA nicht in der Lage ist, im Zentrum der Doppelschicht zu verbleiben. Die Fähigkeit von SM-102 und ALC-0315, die POPC-Membran zu durchdringen, kann mit ihrer Sättigung im Vergleich zu DLin-MC3-DMA zusammenhängen.“
“Vergleicht man DLin-MC3-DMA mit SM-102 und ALC-0315 in Gegenwart der POPC-Lipiddoppelschicht (siehe Abb. 2(b)), so lässt sich feststellen, dass ALC-0315 die höchste Affinität zur Membran aufweist, während DLin-MC3-DMA die niedrigste hat. Sowohl ALC-0315 als auch SM-102 können sich problemlos in der Mitte der Lipiddoppelschicht festsetzen. Dies steht auch im Einklang mit den in Tabelle 1 dargestellten Daten.”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9250827/

Ihr erinnert euch an mein Anfangsstatement dass wir von Fettkugelbomben reden?
Hier also der Grund, wieso ich pessimistisch bin, dass die RNA-Expressionsraten, die gemessen wurden für die Biodistribution etwas darüber aussagen werden, wieviele Zellen tatsächlich effektiv transfiziert wurden.
Wie Ermilova und Swenson festhalten, kann sich ALC-0315 problemlos in phospholipiden Doppelschichten verankern. Was wir mit Sicherheit sagen können: Es sind bestimmte Lipid-Verhältnisse und es gibt keine Garantie, dass diese sich nicht während der Transfektion verändern würden und ein Teil der Lipide sich einfach in die Membran integrieren würde. Denn wir reden hier nicht mehr von Endozytose sondern Penetration. Sprich einem Gewaltakt. Noch dazu halten die Autoren weiter fest:
“Wir entdeckten, dass DLin-MC3-DMA eine Affinität zu der am stärksten gesättigten Monokomponenten-Lipiddoppelschicht 1,2-Dimyristoyl-sn-Glycero-3-Phosphocholin (DMPC) und eine Abneigung gegen das ungesättigte 1,2-Dioleoyl-sn-Glycero-3-Phosphocholin (DOPC) aufweist. Die Vorliebe für eine bestimmte Membran war auch gut mit den Phasenübergangstemperaturen von Phospholipid-Doppelschichten sowie mit ihren strukturellen und dynamischen Eigenschaften korreliert. Außerdem durchdrang das ionisierbare Lipid im Falle der Anwesenheit von DLin-MC3-DMA in der Membran mit DOPC diese, was auf mögliche synergistische Effekte hinweist [Eine Wechselwirkung, durch die Ergebnisse (Effekte) multipliziert werden.].”
Doppelouch. Das heißt also, es käme nicht mal unbedingt auf den Zelltypus an, sondern auf die Membransättigung während des Transfektionsmomentums. Wir wissen ja, dass phospholipide Doppelschichten in permanenter Bewegung sind.
https://www.nature.com/articles/s41467-020-14528-1

“Zellen erhalten die Membranfluidität aufrecht, indem sie die Lipidsättigung regulieren, aber die molekularen Mechanismen dieser homöoviszischen [gleichartigen] Anpassung sind noch wenig bekannt. Wir haben die zentrale Maschinerie zur Regulierung der Lipidsättigung in der Bäckerhefe rekonstruiert, um ihren molekularen Mechanismus zu untersuchen. Durch die Kombination von Molekulardynamiksimulationen mit Experimenten konnten wir eine bemerkenswerte Empfindlichkeit des Transkriptionsregulators Mga2 gegenüber der Häufigkeit, Position und Konfiguration von Doppelbindungen in Lipid-Acylketten aufdecken und Einblicke in die molekularen Regeln der Membrananpassung gewinnen. Unsere Daten stellen die vorherrschende Hypothese in Frage, dass die Membranfluidität als Messgröße für die Regulierung der Lipidsättigung dient. Vielmehr zeigen wir, dass Mga2 die molekulare Lipid-Packungsdichte in einer bestimmten Region der Membran erfasst. Unsere Ergebnisse deuten darauf hin, dass Sensoren für Membraneigenschaften eine bemerkenswerte Empfindlichkeit für hochspezifische Aspekte der Membranstruktur und -dynamik entwickelt haben, was den Weg für die Entwicklung genetisch kodierter Reporter für solche Eigenschaften in der Zukunft ebnet.”
Und:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6178887/
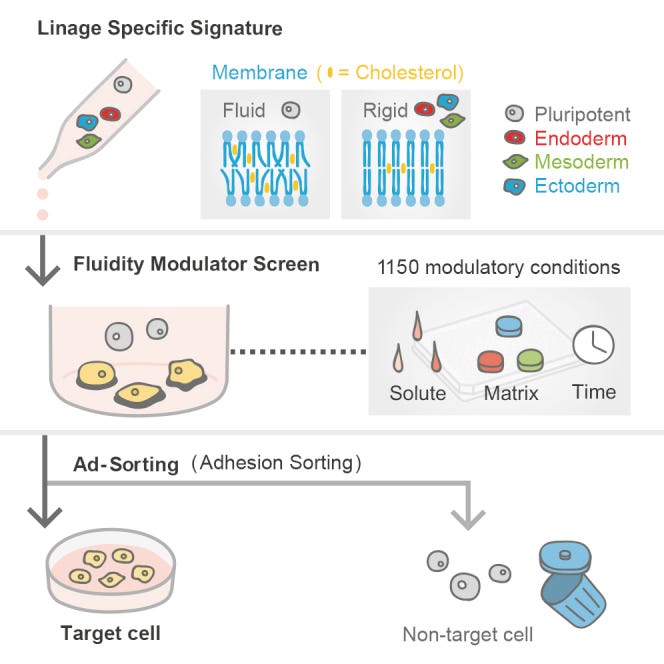
”Die Fluidität der Zellmembran ist ein entscheidender Modulator der Zelladhäsion und -migration, was uns dazu veranlasst hat, die systematische Landschaft der linienspezifischen zellulären Fluidität während der Differenzierung zu definieren. Hier haben wir die Landschaft der Membranfluidität in verschiedenen Abstammungslinien - von der menschlichen Pluripotenz bis zu differenzierten Nachkommen - aufgedeckt: (1) die Versteifung der Membran geht dem Austritt aus der Pluripotenz voraus, (2) die Zusammensetzung der Membran moduliert die Übertragung von Aktivin-Signalen, und (3) die Signaturen sind relativ keimschichtspezifisch, vermutlich aufgrund der einzigartigen Lipidzusammensetzung. Durch die Modulation der variablen linienspezifischen Fluidität haben wir eine markierungsfreie "Adhäsionssortierungsmethode (AdSort)" mit einfacher kultureller Manipulation entwickelt, die pluripotente Stammzellen effektiv eliminiert und die Zielpopulation als Ergebnis der mehr als 1.150 untersuchten Bedingungen, bei denen Verbindungen und Matrices kombiniert wurden, reinigt. Diese Ergebnisse unterstreichen die wichtige Rolle der einstellbaren Membranfluidität bei der Beeinflussung der Aufrechterhaltung und Differenzierung von Stammzellen, die in eine linienspezifische Zellreinigungsstrategie umgesetzt werden kann.”
Also so wie ich das Ganze jetzt sehe - sofern ich hier keinen Logikfehler mache - ,würde jederzeit noch die Möglichkeit für mehr Transfektionsmomente bestehen, solange diese LNPs im System rumwabern. Bye, bye, Bluthirnschranke. Und alleine die Aussage, dass nicht mal die Mechanismen für die Fluiditätsregeln richtig erforscht sind und ich hier zwei kontroverse Thesen zitiere, lässt mich erneut zusammenzucken.
Dies noch mal bewusst gemacht kommen wir endlich zur Genexrpessionsalteration:

“Die leeren Lipid-Nanopartikel des BNT162b2-Impfstoffs sind in der Lage, eine NF-κB-Reaktion auszulösen
Die NF-κB-Antwort war vergleichbar mit derjenigen auf niedrige Dosen von R848, einem TLR-7/8-Agonisten, sowie auf LPS, einem TLR4-Agonisten [hier: rezeptorbindend]. Als Nächstes untersuchten wir die NF-κB-Aktivierung in Abwesenheit von MyD88 oder TRIF anhand von Knockout-Reporterzelllinien. Wir konnten zeigen, dass die NF-κB-Aktivierung in beiden Knockout-Zelllinien reduziert ist. Im Vergleich dazu stimulierte LPS die NF-kB-Aktivierung in erster Linie durch MyD88, während R848 auf beide Wege angewiesen ist.
Unsere Daten zeigen, dass LNPs wichtige angeborene Immunzellen über den NF-kB-Weg, nicht aber über IRF [ein weiterer Transcriptionsfaktor] stimulieren können, was darauf hindeutet, dass dieser Hauptregulator an der durch die aktuelle mRNA-Impfstoffplattform ausgelösten Immunaktivierung beteiligt sein könnte. Darüber hinaus deuten unsere Daten darauf hin, dass LNPs sowohl die MyD88- als auch die TRIF-Signalkaskaden nutzen können, um eine Aktivierung auszulösen. Das Verständnis der Mechanismen, die hinter der immunstimulierenden Kapazität von LNPs stehen, kann Licht auf die Modulation dieser Impfstoffkomponenten werfen, um das Impfstoffdesign zu verbessern.”
Leider gibt es nur das Abstrakt dieser vom Defense Health Program finanzierten Studie. Doch hier wird bereits klar, dass die Signalkaskaden NF-kB getriggert und Genexpressionsalterationen damit zwingend ausgelöst haben müssen. Welche, wollte man vermutlich nicht verraten, da man sich ansonsten hätte entblößen müssen und zeigen, dass es eben nicht die erwünschte Spikeantwort war, die als sog. Immunantwort ausgelöst wurde. Doch es sollte wahrhaft niemanden mehr überraschen, denn es gab mittlerweile genügend Publikationen, die diese Beobachtung bestätigen.
Kommen wir damit (endlich) zu meinem absoluten Lieblingspaper - Ndeupen et al.:
https://www.cell.com/iscience/fulltext/S2589-0042(21)01450-4

“Impfstoffe auf der Grundlage von mRNA-haltigen Lipid-Nanopartikeln (LNPs) sind eine vielversprechende neue Plattform, die von zwei führenden Impfstoffen gegen COVID-19 verwendet wird. [Blabla, bla, BLA] Klinische Studien und laufende Impfungen weisen unterschiedliche Schutzniveaus und Nebenwirkungen auf. Die Ursachen für die gemeldeten Nebenwirkungen sind jedoch nach wie vor unklar. Hier präsentieren wir Beweise dafür, dass die LNPs von Acuitas, die in präklinischen Studien mit nukleosidmodifizierten mRNA-Impfstoffen verwendet werden, bei Mäusen stark entzündlich wirken. Die intradermale und intramuskuläre Injektion dieser LNPs führte zu schnellen und robusten Entzündungsreaktionen, die durch massive Neutrophileninfiltration, Aktivierung verschiedener Entzündungswege und Produktion verschiedener entzündlicher Zytokine und Chemokine gekennzeichnet waren. Die gleiche Dosis von LNP, die intranasal verabreicht wurde, führte zu ähnlichen Entzündungsreaktionen in der Lunge und hatte eine hohe Sterblichkeitsrate zur Folge, wobei der Mechanismus nicht geklärt ist. Die Wirksamkeit der mRNA-LNP-Plattformen bei der Induktion adaptiver Immunreaktionen und die beobachteten Nebenwirkungen könnten also auf die stark entzündliche Natur der LNP zurückzuführen sein.”
DER Mechanismus, wenn es zig wären? Die Ursachen diskutieren wir ja gerade.
Figur F. Ich markierte einige Signale, bei denen sich mir der Magen rumdreht (NES = normalized enrichment score)

Ich könnte nun für jeden falsch regulierten Teil hier noch jede Menge Papers mit übelstem Ausgang rauskramen. Aber ich denke, das würde gerade endgültig den Rahmen sprengen.
Und weiter heißt es in dieser wirklich brillanten und gnadenlos detaillierten Analyse:
“Mit p < 0,05 und FDR<0,05 wurden 9.508 Gene bzw. 8.883 Gene unterschiedlich exprimiert. Noch wichtiger ist, dass die Gene, die mit der Entwicklung, Rekrutierung und Funktion von Monozyten/Granulozyten (Cxcl1, Cxcl2, Cxcl5, Cxcl10, Ccl2, Ccl3, Ccl4, Ccl7, Ccl12, Csf2 und Csf3) und mit Entzündungen (Il1b und Il6) in Verbindung gebracht werden, im Vergleich zu den Kontrollproben die höchsten Steigerungen aufwiesen (Abbildung 2E). Wir beobachteten auch eine signifikante Hochregulierung von Gentranskripten, die mit der Aktivierung von Inflammasomen in Verbindung gebracht werden, wie z. B. Il1b und Nlrp3, und eine Herabregulierung von Nlrp10, von dem bekannt ist, dass es Inflammasome hemmt (Abbildung 2E). Gensatzanreicherungsanalysen (GSEA) zeigten die Aktivierung vieler verschiedener Entzündungswege, einschließlich, aber nicht beschränkt auf Virusinfektionen, RIG-I, NOD-like und Toll-like-Rezeptor-Signalisierung (Abbildung 2F). Pro-apoptotische und nekroptotische Gensätze wurden ebenfalls signifikant hochreguliert, ebenso wie die Interferonsignalisierung (Abbildung 2F).”
Dies wurde auch - in nicht ganz so umfangreichen Ausmaß - durch folgendes Paper bestätigt.
Es wurden hier jedoch andere ionisierte Lipidformulierungen verwendet. Dennoch ist es mehr als ausreichend um meine Ausgangsaussage, dass sämtliche Signalwege in den transfizierten Zellen alteriert sind, zu unterstreichen. Denn wie wir ja nun zusammen lernten und ich hoffentlich halbwegs verständlich erklären konnte, wird es ohne Signaltransduktion keine Genexpressionsalterationen in einer Zelle geben.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9936473/

“Wir untersuchten auch die Produktion von Zytokinen nach der in vitro DC-Reifung durch eLNP. IL-6 (p = 0,0321), IL-12 (p = 0,0294), IL-21 (p < 0,0001), CD40L (p = 0,0009), IFNα (p = 0,0040) und IFNγ (p = 0,0008) stiegen nach 24 Stunden eLNP-Stimulation deutlich an (Abb. 1b). Somit kann eLNP sowohl pro-TFH-Zytokine als auch wichtige Zytokine und Chemokine induzieren, die bei der Aktivierung der angeborenen Immunantwort wie IFN-α und IFN-γ wirksam sind.”
Was all diese Autoren jedoch nicht zugeben werden: Was auch immer ihr also nach der Transfektion an Antikörpern und aktivierten Immunzellen triggert und ausspuckt hat nichts und ich meine wirklich gar nichts mehr mit einer nat. Immunreaktion zu tun, sondern ist eine zur Gänze umprogrammierte Reaktion. Und hier kommt dann eben der echte Brüller: Ihr wisst hoffentlich, dass ihr in jeder der Trillionen von Zellen die gleiche Anzahl an Genen habt, einige sind ausgeschaltet, andere gedimmt, wieder andere werden aktiviert, wenn sie gebraucht werden. Doch diese Regeln sind von Zelltyp zu Zelltyp und von Organ zu Organ absolut einmalig. Was könnte da schon bei Genexpressionsalterationen in “Target unknown” schief gehen?
Die Clotshots und was so alles schiefgehen kann: The good, the bad and the transfected

https://portlandpress.com/DocumentLibrary/Umbrella/Cell%20Signaling/csb0001012.full.pdf

“Eine Vielzahl von Krankheiten wird durch Defekte in Signalwegen verursacht. Die Art dieser Defekte und die Art und Weise, wie sie hervorgerufen werden, sind sehr unterschiedlich. Einige dieser Defekte können durch pathogene Organismen und Viren verursacht werden, von denen viele in die Signalvorgänge eingreifen können. Andere Krankheiten lassen sich auf Defekte in der Funktion von Zellsignalübertragungswegen zurückführen. Das Konzept der Umgestaltung von Signalsomen und Krankheiten bietet einen Rahmen für die Betrachtung, wie Defekte in Signalwegen zu Krankheiten führen können. Es ist zweckmäßig, diese Defekte in phänotypische Umgestaltung des Signalsoms und genotypische Umgestaltung des Signalsoms zu unterteilen. Die meisten schweren Krankheiten beim Menschen, wie Bluthochdruck, Herzkrankheiten, Diabetes und viele Formen psychischer Erkrankungen, scheinen auf subtile phänotypische Veränderungen der Signalwege zurückzuführen zu sein. Eine solche phänotypische Umgestaltung verändert das Verhalten von Zellen, so dass ihre normalen Funktionen untergraben werden, was zu Krankheiten führt. Da es sich als schwierig erwiesen hat, diesen Zusammenhang zwischen der Umgestaltung von Signalsomen und Krankheiten eindeutig festzustellen, wurden bei der Entwicklung wirksamer Behandlungen nur relativ geringe Fortschritte erzielt.”
Dieses Paper ist genial und äußerst umfangreich und erklärt anhand von Altzheimer und Parkinson, wie die Signalwege affektiert sind. Leider ist es zu umfangreich, um es in Gänze zu diskutieren. Wichtig fand ich jedoch noch folgenden Passus zu zitieren:
“Entzündungen und Alterung
Die Alterung geht häufig mit dem Auftreten eines schwach ausgeprägten Entzündungsmilieus einher. Diese Entzündungsreaktion kann entweder durch die Aktivierung der angeborenen oder der adaptiven Immunantwort ausgelöst werden. Was genau diese Entzündungsreaktionen in normal alternden Geweben auslöst, ist nicht sofort klar. Diese Entzündung, die in der Regel durch ansässige Makrophagen in peripheren Geweben oder die Mikroglia im Gehirn ausgelöst wird, ist mit der Bildung einer "Entzündungssuppe" verbunden, die viele entzündungsfördernde Mediatoren und toxische Faktoren wie reaktive Sauerstoffspezies (ROS) enthält, die direkt in die verschiedenen Mechanismen einfließen, die zum Alterungsprozess beitragen (Modul 12: Abbildung Alterungsmechanismen). Die pro-inflammatorischen Mediatoren wie der Tumornekrosefaktor α (TNFα) und Zytokine wie Interleukin-1 wirken über die TNFα-Rezeptoren bzw. Toll-like-Rezeptoren (TLRs), um Informationen entweder über den Nuklearfaktor κB (NF-κB) oder den MAPK-Signalweg (p38 und JNK) weiterzuleiten. Diese niedriggradige Entzündung ist nicht immer schädlich, sondern kann oft sogar von Vorteil sein, da eine wechselseitige Kommunikation zwischen Immunzellen und benachbarten Gewebezellen oft von Vorteil ist, da sie zur Aufrechterhaltung der phänotypischen Stabilität beitragen kann. Ein gutes Beispiel für diesen zellulären Dialog sind die vielfältigen Wirkungen des Nuklearfaktor-κB-Signalwegs (NF-κB), der die Expression sowohl von proinflammatorischen Zytokinen als auch von Immunregulatoren fördern kann (Modul 2: Abbildung Toll-Rezeptor-Signalweg). Ein weiteres Beispiel findet sich im Gehirn, wo das entzündliche Zytokin Tumornekrosefaktor α (TNFα) die Expression von Signalkomponenten wie dem Inositol-1,4,5-Trisphosphat (InsP3)-Rezeptor und dem anti-apoptotischen Protein Bcl-2 aufrechterhalten kann. Dieses feine Gleichgewicht zwischen der positiven und der negativen Wirkung von Entzündungen beginnt sich bei der Entstehung verschiedener Krankheiten in Richtung der negativen Wirkung zu verschieben.”
Na? Verwirrt, wieso die Autoren hier die P38 MAPk und aktiviertes NF-κB als separierte Signalwege behandeln? Müsst ihr nicht: Viele Wege führen zum Nuclearfaktor Kappa B und das Blöde ist, kaum jemand hat sich wirklich mit der Mannigfalitgkeit und den Crosstalks beschäftigt. Und ich sagte ja schon: Raum-Zeit-Interaktion.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2701546/

Wie ich ja oben angekündigt habe, wollte ich noch kurz das P38 anreißen um zu zeigen, was so schief gehen kann, wenn man die Signalkaskaden falsch taktet und anders ansteuert, als vorgesehen, wie in der Publikation auch wunderschön detailliert gezeigt.

“Der Thrombozytenfaktor 4 (PF4) ist ein reichlich vorhandenes Chemokin, das bei Aktivierung aus den α-Granula der Thrombozyten freigesetzt wird. PF4 spielt eine zentrale Rolle in der Pathophysiologie der impfstoffinduzierten Immunthrombozytopenie und -thrombose (VITT), bei der Antikörper gegen PF4 Immunkomplexe mit PF4 bilden, die über Fc-Rezeptoren Thrombozyten und Neutrophile aktivieren. In dieser Studie zeigen wir, dass PF4 den Thrombopoietin-Rezeptor, das zelluläre myeloproliferative Leukämieprotein (c-Mpl), auf Blutplättchen bindet und aktiviert. Dies führt zur Aktivierung der Janus-Kinase 2 (JAK2) und zur Phosphorylierung von Signal Transducer and Activator of Transcription (STAT3) und STAT5, was zur Thrombozytenaggregation führt. Die Hemmung des c-Mpl-JAK2-Signalwegs hemmt die Thrombozytenaggregation gegenüber PF4, VITT-Seren und der Kombination aus PF4 und IgG, das aus dem Plasma von VITT-Patienten isoliert wurde. Die Ergebnisse unterstützen ein Modell, bei dem PF4-basierte Immunkomplexe Thrombozyten durch Bindung der Fc-Domäne an FcγRIIA und PF4 an c-Mpl aktivieren.”
Und weiter heißt es:
”Diese Ergebnisse zeigen, dass hohe Konzentrationen von PF4 (>10 μg/ml) über den TPO-Rezeptor c-Mpl eine robuste Thrombozytenaggregation auslösen können und dass niedrigere Konzentrationen die Thrombozytenaktivierung über denselben Weg verstärken können. In Thrombozyten signalisiert c-Mpl über die Tyrosinkinase JAK2, die stromaufwärts von STAT3 und STAT5a/b liegt, sowie über mehrere andere Signalwege (nämlich p38-Mitogen-aktivierte Proteinkinase, extrazelluläre signalregulierte Kinase-cytosolische Phospholipase A2 [ERK2-cPLA2] und Phosphatidylinositol-3-Kinase). Der relative Beitrag jedes dieser Signalwege zur Thrombozytenaktivierung durch PF4 ist nicht bekannt.”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5438729/

Für den nächsten Gedanken, was so alles an den gängigsten Rezeptoren schief ging, brauche ich kurzen Vorlauf um eine bösartige Frage einfach mal so in den Raum zu schmeißen: Denkt eigentlich noch irgendwer, dass es keine epigenetischen Folgen nach der Transfektion gab?
https://www.nature.com/articles/s41467-021-25728-8

https://pubmed.ncbi.nlm.nih.gov/28853098/

Ich mein ja nur mal: Wenn also die MAPk via Inhibition den zytosolischen RIG-1-Like-Pattern-Erkennungs-Rezeptor epigenetisch umgestaltet? Und wenn wir dann einen Blick in Föhse et al. werfen, welche endlich - nach Entschärfung der Formulierungen im Abstrakt und der Überschrift - peer reviewed wurden, erscheint die Frage doch recht berechtigt:
sciencedirect.com/science/article/pii/S1521661623005259

“Interessanterweise zeigte die RNA-Sequenzierung langfristige Veränderungen in den Transkriptionsprogrammen von Immunzellen nach Verabreichung des BNT162b2-Impfstoffs, und die Impfung modulierte auch die Produktion von Entzündungszytokinen nach Stimulation mit viralen, bakteriellen und fungialen Stimuli. Die Synthese und Freisetzung von myeloiden Zytokinen aus dem IL-1/IL-6-Signalweg war sechs Monate nach der ersten Dosis von BNT162b2 tendenziell höher. Im Gegensatz dazu nahm die Produktion von IFN-α nach Stimulation mit SARS-CoV-2, dem TLR3-Liganden Poly I:C und dem TLR7/8-Liganden R848 nach der Impfung ab. Insgesamt konnten wir beobachten, dass die Verabreichung des BNT162b2-Impfstoffs die angeborene Immunantwort bis zu einem Jahr nach der ersten Impfung modulierte.”
Und das heißt nicht zwingend, dass es nach einem Jahr besser geworden wäre, sondern nur, dass sie ein Jahr beobachteten. Erste DOSIS! (“bis zu einem Jahr nach der ersten Impfung modulierte.”)
”Die Proben wurden entsprechend der von BioNTech und Pfizer durchgeführten Phase-1-Studie [1] zu fünf Zeitpunkten entnommen: vor der Impfung (t0), drei Wochen nach der ersten Dosis von 30 μg BNT162b2 (t1), zwei Wochen nach der zweiten Dosis (t2) - d. h. fünf Wochen nach der ersten Dosis, sechs Monate nach der ersten Dosis (t3) und vier Wochen nach der Auffrischungsimpfung, die etwa ein Jahr nach der ersten Dosis erfolgte (t4), um einen umfassenderen Überblick über mögliche Langzeitwirkungen der Impfung zu erhalten.”
Wollen wir ein Spiel spielen und mal kurz fragen, wo der Rest der Auswertung nach 2. und 3. Dosis wären?
Erinnert ihr euch, dass ich weiter oben schon anmerkte, dass die MAPk auch in der Regulierung des TLR-Netzwerkes (alle unter einander in Crosstalks [PLURAL]) auch eine essentielle Rolle spielt… Ich mein ja nur mal.
Die Studie von Föhse et al. deckt sich dann bedauerlicherweise auch mit der Observierung bei Kindern.
https://frontiersin.org/articles/10.3389/fimmu.2023.1242380/full


Aber hey: Keine Panik! Die Autoren waren begeistert und empfanden es nicht als notwendig auch nur im Ansatz eine kritische Frage aufzuwerfen. Ganz im Gegenteil. Die Autoren feierten es als adäquate Immunantwort. Wenn dem doch so ist: Wieso vermieden sie es, wie der Teufel das Weihwasser mit einer Gesunden und einer Genesenen Kontrollgruppe zu vergleichen? Noch dazu gab es nicht einen einzigen schweren oder gar tödlichen Verlauf unter ‼gesunden‼ Kindern. Gesunde Kinder: Ihr wisst schon, diese süßen Geschöpfe mit den triefenden Nasen zumeist schmutzigen Händen, lebendigen Augen und herzerwärmendem Gelächter.
https://twitter.com/RetsefL/status/1638894943308599297

https://link.springer.com/article/10.1007/s00431-022-04587-5

Um die Aussage faktisch zu untermauern.
Doch zurück zum Thema:
Ich rekapituliere noch mal: Ich habe mit euch also jetzt soweit die Entstehung von Erstsignalen downstream nach Transfektion diskutiert. Wir haben geschaut, wie diese auch bei der Membranreorganisation beteiligt sind und die Membran eben jene Signale steuert und auch die gesamten Signalrezeptoren, wie VEGFR, TGFR, TLR und EGFR strukturiert und dadurch auch die Funktionalität organisiert. Wir diskutierten, was die Überexpressierung und Aktivierung eines einzigen Signalproteins schon als Folge haben kann und das “die Experten”™ scheinbar viel zu wenig Signaltransduktionswissenschaft studierten oder in böser Absicht täuschen wollen und handeln.

Houston? Vielleicht haben wir ein Problem: Je überexpressierter PD-L1, desto härter wird es für eure CD8+ T-Zellen.
https://www.degruyter.com/document/doi/10.1515/cclm-2022-0787/html?lang=de

Papertitel:
Increased PD-L1 surface expression on peripheral blood granulocytes and monocytes after vaccination with SARS-CoV2 mRNA or vector vaccine
Sollen wir noch ein Review kurz anreißen, um die Katastrophe für Immunsystem und garantierte Krebszunahme zu begreifen?
https://www.nature.com/articles/s41392-021-00658-5

“Wir erörtern die Entstehung und Beendigung von Entzündungen sowie die Wechselwirkungen zwischen Tumorentwicklung und Entzündungsprozessen.”
Eigentlich reicht folgender Passus:
”Der Zusammenhang zwischen Entzündungen und Krebs wurde erstmals Mitte des 19. Jahrhunderts von Rudolf Virchow vorgeschlagen, der beobachtete, dass Krebs an Orten chronischer Entzündungen entsteht und dass Entzündungszellen in Tumorbiopsien reichlich vorhanden sind. Heutzutage gelten krebsbedingte Entzündungen als ein Hauptmerkmal von Krebs, wobei ein Zusammenhang zwischen chronischen Entzündungen und der Tumorentwicklung gut belegt ist. Tatsächlich werden chronische, dysregulierte, anhaltende und ungelöste Entzündungen bei den meisten Krebsarten mit einem erhöhten Risiko für bösartige Erkrankungen sowie mit dem bösartigen Fortschreiten von Krebs in Verbindung gebracht. Darüber hinaus gibt es immer mehr Hinweise darauf, dass die entzündliche Mikroumgebung des Tumors (TME) ein entscheidender Faktor für die therapeutische Wirksamkeit der konventionellen Chemotherapie (z. B. Strahlen- und Chemotherapie) und der Immuntherapie ist. Es wurde jedoch berichtet, dass eine durch exogene Stimulatoren ausgelöste akute Entzündung die Anti-Tumor-Immunität verstärkt, indem sie die Reifung und Funktion von dendritischen Zellen (DCs) und die Initiierung von Effektor-T-Zellen fördert.
Entzündungen, an denen das angeborene und das adaptive Immunsystem beteiligt sind, sind bekanntlich die schützende Immunreaktion zur Aufrechterhaltung der Gewebehomöostase durch die Beseitigung schädlicher Reize, einschließlich geschädigter Zellen, Reizstoffe, Krankheitserreger und steriler Läsionen. Im Gegensatz zur Wundheilung und Infektion ist die Entzündungsreaktion während der Krebsentwicklung nachweislich nicht auflösend. Darüber hinaus ist bekannt, dass tumorexterne Entzündungen durch verschiedene Faktoren ausgelöst werden, darunter Autoimmunerkrankungen, bakterielle und virale Infektionen, Fettleibigkeit, Rauchen, Asbestexposition und übermäßiger Alkoholkonsum, die alle das Krebsrisiko erhöhen und das Fortschreiten der Krebserkrankung beschleunigen. Im Gegensatz dazu können krebsimmanente oder krebsauslösende Entzündungen durch krebsauslösende Mutationen verursacht werden und durch die Rekrutierung und Aktivierung von Entzündungszellen zur Tumorprogression beitragen. Es ist bekannt, dass sowohl extrinsische als auch intrinsische Entzündungen zu einem immunsuppressiven TME führen und damit eine bevorzugte Bedingung für die Tumorentwicklung darstellen. Sobald das entzündliche TME etabliert ist, würden Entzündungsfaktoren, die von Tumorzellen oder interstitiellen Zellen stammen, die Zellproliferation induzieren und das Zellüberleben verlängern, indem sie zunächst Onkogene aktivieren und anschließend Tumorsuppressorgene inaktivieren.”
Aber gut. Wir wollen es ja detailliert betrachten:
”Die NOD-ähnlichen Rezeptoren sind Mustererkennungsrezeptoren im Zytoplasma. Die strukturellen Merkmale der NLRs sind folgende: die zentrale nukleotidbindende Oligomerisierungsregion (NACHT), die für die Oligomerisierung und Aktivierung der NLRs sehr wichtig ist, ist eine gemeinsame Struktur der NLR-Familie; die N-terminale Effektor-Bindungsregion, d. h. die N-terminale Protein-Protein-Interaktionsdomäne, wie z. B. die Caspase-Aktivierungs- und Rekrutierungsdomäne (CARD); und die C-terminale Anreicherung leucinhaltiger Wiederholungen (LRRs). Die NLR-Familie besteht aus 22 Arten intrazellulärer Mustererkennungsmoleküle, die in einer Vielzahl von Gewebezellen verteilt sind, darunter Monozyten, Makrophagen, T-Zellen, B-Zellen, dendritenähnliche Zellen des Dünndarms und Paneth-Zellen. Menschliche NLRs werden in die folgenden 5 Kategorien unterteilt: NLRA, NLRB, NLRC, NLRP und NLRX. Es wurde gezeigt, dass NOD1 und NOD2 durch CARD-CARD-Interaktionen [ganz komplizierte NOD-Like-Rezeptorfunktions-Details, die man nicht unbedingt braucht, um zu begreifen, dass die NOD-Like-Rezeptoren also ebenfalls essentielle Rollen spielen] das rezeptor-interagierende Protein (RIP)-2 rekrutieren und dadurch die NF-κB- und mitogen-aktivierten Proteinkinase (MAPK)-Signalpfade aktivieren. Die Kombination des PYD-haltigen NLRP-Proteins und des CARD-haltigen Apoptose-assoziierten Speck-ähnlichen (ASC) Proteins führt nachweislich zur Aktivierung von Caspase-1 und fördert damit eine Entzündungsreaktion.”
NOD-Like-Rezeptoren sind ebenfalls zytosolische Pattern-Erkennungsrezeptoren. Und wie man aus der vorherigen Publikation, die die Umfunktionalisierung der Immunantwort als Erfolg feiert, entnehmen kann, waren auch diese essentiell involviert. Tja… Ich will ja nicht sagen, dass die Transfektionsschüsse ne Krebsbombe sind, aber es sind verfluchte Krebsbomben!
Apropos chronische Entzündungen… War da nicht noch irgendwas mit den IgG4-Klassenswitches, welche auf chronische Entzündung hinweisen?:
https://www.nature.com/articles/s41598-023-40103-x

https://www.science.org/doi/10.1126/sciimmunol.ade2798
https://www.journalofinfection.com/article/S0163-4453(24)00053-7/fulltext
https://www.mdpi.com/2076-393X/11/5/991
https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2022.1020844/full
https://www.biorxiv.org/content/10.1101/2023.09.15.557929v2
https://www.jstage.jst.go.jp/article/internalmedicine/62/10/62_1125-22/_article
https://www.ijidonline.com/article/S1201-9712(23)00789-0/fulltext
https://www.mdpi.com/2076-393X/11/11/1720
Aber gut. Sind ja nur 9 Publikationen mit Vorankündigung seit 2021, als wir wussten, dass es JAK2 zerklumpt und Interleukin (Il)-6 überexpressiert wird. Muss also cherrygepickt sein, weil “die Experten”™ nichts sagten/ sagen.
Und was die T-Zellen angeht:
https://www.science.org/doi/10.1126/science.abm0829

https://sciencedirect.com/science/article/pii/S1286457920300721

https://ncbi.nlm.nih.gov/pmc/articles/PMC8879995/

Sorry. Sieht jetzt nicht so rosig aus, liebe Impffaschisten. Ihr habt euch zunächst mal die TH1/TH2/TH17-Verhältnisse zerballert und so wie es aussieht, rauchen euch die gespikten T-Zellen nach 6 Monaten eventuell weg - was eigentlich mit den Antikörpertitern geschehen sollte, da wir von einem Sarbecovirus reden. Und genau solche Falschregulierungen kommen Upstream dabei rum, wenn man downstream richtig Scheiße baut.
Poah. Mir raucht der Schädel, mein Magen dreht sich rum und ich habe endgültig schlechte Laune. Nichtsdestotrotz hoffe ich, ihr konntet bis hierhin folgen. Wenn ihr das konntet, sollte euch folgendes Zitat aus der ersten Publikation von W. Dörfler, welche er danach ganz fix mit einer 2. Publikation relativierte, nicht mehr überraschen:
https://ncbi.nlm.nih.gov/pmc/articles/PMC8168329/

“Die menschliche Bevölkerung ist gegenwärtig in einem riesigen Experiment fremder DNA ausgesetzt. Nach Abschluss der weltweiten Impfungen sollte ein Sentinel-Programm für die Zeit nach der Impfung eingerichtet werden, um die Verschlimmerung unerwarteter, möglicherweise neuartiger menschlicher Krankheiten bei geimpften Personen zu überwachen.”
Was ich noch fix anmerken muss: Wir wissen nicht, inwieweit sich irgendwas von den falsch getriggerten Signalen eventuell erholen könnte und es gibt einige Linderungsansätze für die schlimmsten Symptome und Effekte. Es könnte genauso gut vorrangig Menschen mit Mutationen in beispielsweise den RAF, RAS und BRCA1/2-Genen betreffen (vielen Dank, lieber Jikkyleaks für diesen wichtigen Hinweis). Dennoch denke und befürchte ich, ist da eine solche Sprengkraft in den Transfektionsschüssen, dass allerspätestens mit der 3. Transfektion ein unumkehrbarer Schaden angerichtet wurde, den ihr euch als tickende Zeitbombe vorstellen müsst. Wenn die Spikes und klebrige modRNA die Zellen nicht fertig machen, dann diese Kaskaden, die zwar (zumeist) langsame Zahnräder (im Sinne von die Konsequenzen spüren) sind, aber unvorstellbare Konsequenzen auf ALLE erdenklichen Zellen haben werden.
Summieren wir also noch mal die Pathomechanismen der Transfektionsbrühe:
Alterierte Signalkaskaden setzen die transfizierten Zellen in völlig falsche Vorbedingungen
+
modRNA-Simulation wird ausgepackt mit unbekannter Translationsdauer, Rate.
+
Framgeshiftete Junkproteine, welche eventuell amyloide Funktionen ausüben werden.
+
Völlig verrückte Proteinmassen wie 180 - 220 kDa sind mit dabei (wo das Spike nur 141kDa haben sollte und man auf die Prolin-Insertionen + Stopcodonreadthrough mit translatierter 5’UTR tippen könnte)
+
Plasmide Fragmente inklusive einem nukleartransporter mit 72 Basenpaaren
+
Gecleavete Spikes mit aller Pathogenität, die ihr in all den Publikationen lesen könnt
+
Dasselbe Spiel der Signalkaskaden mit der mod-WTF-EVER-IT-IS-RNA-Simulation bei Translation und Faltung und den Spikes, sowie den gebildeten Antikörpern
+
Mögliche LPS/ endotoxische Effekte
+
Alterierte microRNA-Interaktionen
=

Nicht das noch irgendwer auf die Idee käme zu fragen, was denn überhaupt noch von den ursprünglichen Zellfunktionen übrig geblieben wäre. Ich argumentiere damit, dass es beinahe unmöglich für transfizierte Zellen gemacht wurde, noch in einer ordentlichen Apoptose unterzugehen und Seneszenz [Als Seneszenz bezeichnet man das biologische Phänomen, dass die meisten Zellen von Wirbeltieren nach einer bestimmten Zahl von Zellteilungen (Mitosen) in der Zellkultur ihr Wachstum einstellen.] eine der wahrscheinlichsten Zellreaktionen sein wird. Wobei ich hier auch die brillante Zusammenfassung von Dr. Clare Craig als Gegenthese noch fix einwerfen muss.
https://docs.google.com/document/d/1jSyKYhFzZPr_NIbj6h0PRxICqjNBZoTLhCJRqyIktCc/edit#heading=h.skdljx95oyp7

Nichts Genaueres weiß man nicht.
Meiner bescheidenen Meinung nach haben wir spätestens Ende 2020 mit dem Rollout das Feld bekannter biologischer Mechanismen verlassen und können/müssen Zellbiologie komplett neu denken.
Ich hoffe, ihr habt bis hierher durchgehalten und das ein oder andere Informative mitnehmen können. Noch eine klitzekleine persönliche Notiz, wieso ich scheinbar ja doch das ein oder andere Wort in seiner Tragweite begreife und mich ausgerechnet auf so einen Horrortrip einlasse: Ich denke, es liegt a) daran, dass ich sehr viel Zeit zum Lesen habe, b) in einem wahnsinnigen Tempo logische Überschneidungen erkenne und diese versuche zu verbinden und plausibel zu erklären und c) eben weil ich diesen Krams nie studierte, habe ich keinen solchen BIAS, zu denken, dass irgendetwas in Zellbiologie unmöglich wäre und bin einfach nur neugierig wohin mich bestimmte Fragestellungen führen werden. Ok. Mittlerweile hab ich einen klitzekleinen BIAS drin, weil die erste Frage im Kopf ist, die da laut rumpoltert: “Wie spielt die MAPk rein?” Und ich befürchte das Schlimmste für 2030 - 2033. Genau darum gab es halt auch das Konzept von bis zu 10 Jahre LANGZEITstudien, die man ja getrost aus dem Programm strich. Ich bete nur noch dafür, in jedem einzelnen Wort falsch zu liegen und mich zu irren.
Um die Stimmung etwas aufzulockern, gibt es noch einen Rausschmeißer. Eine Parodie auf das Deutsche Impffluencervirus (Wortspiel für Influencer):
Narfige Grüße und wirklich erschöpft nach diesem ellenlangen Höllenritt,
Euer Nerd, äh, Genervter Bürger. [Korrekturlesung by “Hase”]
Genervter - X suspended your account https://twitter.com/NarfGb WTF???
Genervter - I cannot comment on your great X tweets unless you follow me? NARF! https://twitter.com/1NatteringNabob