Downstream Teil 6
Meine erste Folie, mit der ich zufrieden bin und die mir zeitgleich Angst macht.


Ein paar falsch geschaltete Transkriptionsfaktoren und mIRNas werden schon keinen Unterschied machen
In diesem Teil werde ich ganz kurz auf miRNA-Signalgebung (wofür nur sehr wenig Lektüre existiert) eingehen und mich ein wenig näher den Transkriptionsfaktoren widmen (worin downstream Signalkaskaden in der Regel münden um Gene anzusteuern und RNA aus DNA zu transkribieren und zu prozessieren.
Ein weiterer Höllenritt wartet also. Doch mir erscheint es wichtig, bevor ich in meinem letzten Teil endlich das Ganze summieren kann, klare Hinweise zu geben, wie komplex das gesamte Signalnetzwerk ist und dass jede Alteration, die ohne angemessene Metabolismen (beispielsweise Prozessierung im Darm und Vorbereitung für Zellaufnahme) mit Gewalt forciert wurde, fatale Konsequenzen haben kann und vermutlich wird.
Bevor es los geht ‼Achtung‼
Hier wird es sehr viel auch um Immunzellen gehen, da diese wohl scheinbar mit am meisten beobachtet wurden. Die Lektüre ist wirklich sehr beschränkt. Die meisten Papers beschäftigen sich nur mit der Genexpression und den inhibitorischen Effekten der kleinen Powerriesen.
JAK/STAT und micro RNA
“Der Januskinase (JAK)/Signaltransduktor und Aktivator der Transkription (STAT)-Signalweg ist einer der grundlegenden Kommunikationsknotenpunkte der Zellfunktion. Dieser Signalweg ist ein Konglomerat aus Liganden-Rezeptor-Komplexen, den JAK-Tyrosinkinasen (JAK1-3 und Tyrosinkinase-2) und STAT (STAT 1-4, 5a, 5b und 6), DNA-bindenden Familien von Signaltransduktionsproteinen. Der zentrale JAK/STAT-Signalweg umfasst ein schnelles Zytosol-zu-Kern-Signalmodul, das die rasche Weiterleitung extrazellulärer Signale unterstreicht, die von mehr als 50 Zytokinen, Wachstumsfaktoren und stoffwechselrelevanten Hormonen erzeugt werden” (…)
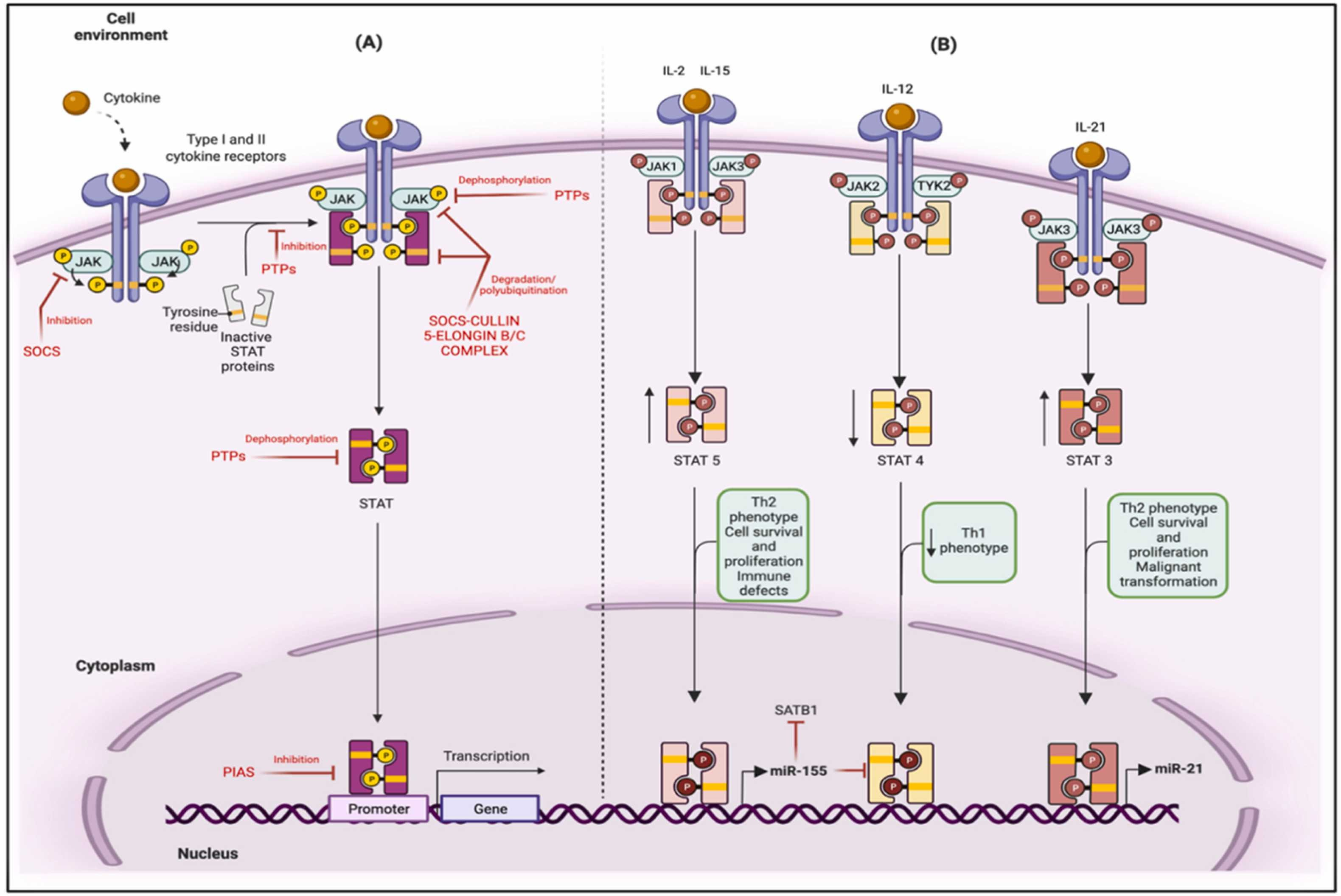
”Durch die konstitutive Aktivierung von STAT5 wird die Expression des onkogenen miR-155 hochreguliert, der die Proliferation und das Überleben bösartiger T-Zellen stimuliert, den Th2-Phänotyp fördert, die Expression des mutmaßlichen Tumorsuppressors SATB1 verringert und deutliche Defekte in der zellulären Immunität hervorruft. Die STAT5-vermittelte Hochregulierung von miR-155 ist auch für den Verlust von STAT4 (der mit der IL-12-Signalgebung verbunden ist) und die Aufhebung von Th1-Reaktionen verantwortlich. Die Signalisierung als Reaktion auf IL-21 (als autokrine Rückkopplungsschleife), die durch die Induktion von STAT3 über konstitutiv aktive JAK3-Kinasen erfolgt, führt zur Hochregulierung von oncomir miR-21, das mit dem Überleben und der Proliferation von Zellen, der malignen Transformation und Th2-Reaktionen in Verbindung gebracht wird.
(TYK2, Tyrosinkinase 2; SH2, Src-Homologie 2; PIAS, Proteininhibitoren aktivierter STATs; PTPs, Protein-Tyrosin-Phosphatasen; CIS, Cytokin-induzierbares SH2-Protein; SOCS, Suppressoren von Cytokin-Signalen; Th1, T-Helfer 1; Th2, T-Helfer 2; IL, Interleukin; und SATB1, spezielles AT-reiches Sequenz-Bindungsprotein 1.)”
NARF! Wer die ganzen Papers noch halbwegs im Kopf hat betreffs TH1/2/17-Verhältnisänderung und Il-21-Expression betreffs IgG4-switches wird hier gerade seine helle Freude haben. [p.S.: Kennt ihr das, in einem Kino als einziger an Stellen zu lachen, die wirklich komisch sind, während ihr bei forcierten Lachmomenten bedeppert dreinschaut?]
Was sich hier dann noch relativ einfach liest, wird ganz schnell ein Alptraum, macht man sich die wirklich komplexe Regulationsaxis, wie eine miR angesteuert wird, bewusst:
STAT5-dependent regulation of CDC25A by miR-16 controls proliferation and differentiation in FLT3-ITD acute myeloid leukemia (STAT5-abhängige Regulierung von CDC25A durch miR-16 kontrolliert Proliferation und Differenzierung bei akuter myeloischer Leukämie mit FLT3-ITD)
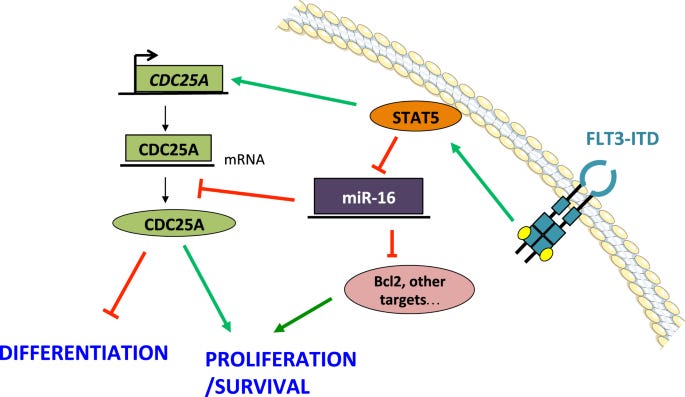
“Schließlich stellte die Verringerung der miR-16-Expression die Proliferation von Zellen, die mit dem FLT3-Inhibitor AC220 behandelt wurden, teilweise wieder her, während die Expression von miR-16-Mimik diese Proliferation stoppte und die monozytäre Differenzierung von AML-Zellen induzierte. Zusammenfassend lässt sich sagen, dass wir eine FLT3-ITD/STAT5/miR-16/CDC25A-Achse identifiziert haben, die für AML-Zellen.” (acute myeloid leukemia)
Perturbed microRNA Expression by Mycobacterium tuberculosis Promotes Macrophage Polarization Leading to Pro-survival Foam Cell

“Die STAT-Familie von Transkriptionsfaktoren spielt eine unterschiedliche Rolle bei der Entwicklung von Makrophagen. miR-1252 und miR-3202 zeigten Komplementarität für STAT1 bzw. STAT2 in ≥2 Datenbanken. Eine Herunterregulierung dieser Transkriptionsfaktoren führt zu einer geringeren Entzündungsreaktion (Tabelle 3).”
Gut, gut: JAK/STAT haben wir also schon mal als nette hübsche Treiber von MicroRNAs identifiziert. Wie sieht es bei mTORC1/2 aus?
mTORC1/2
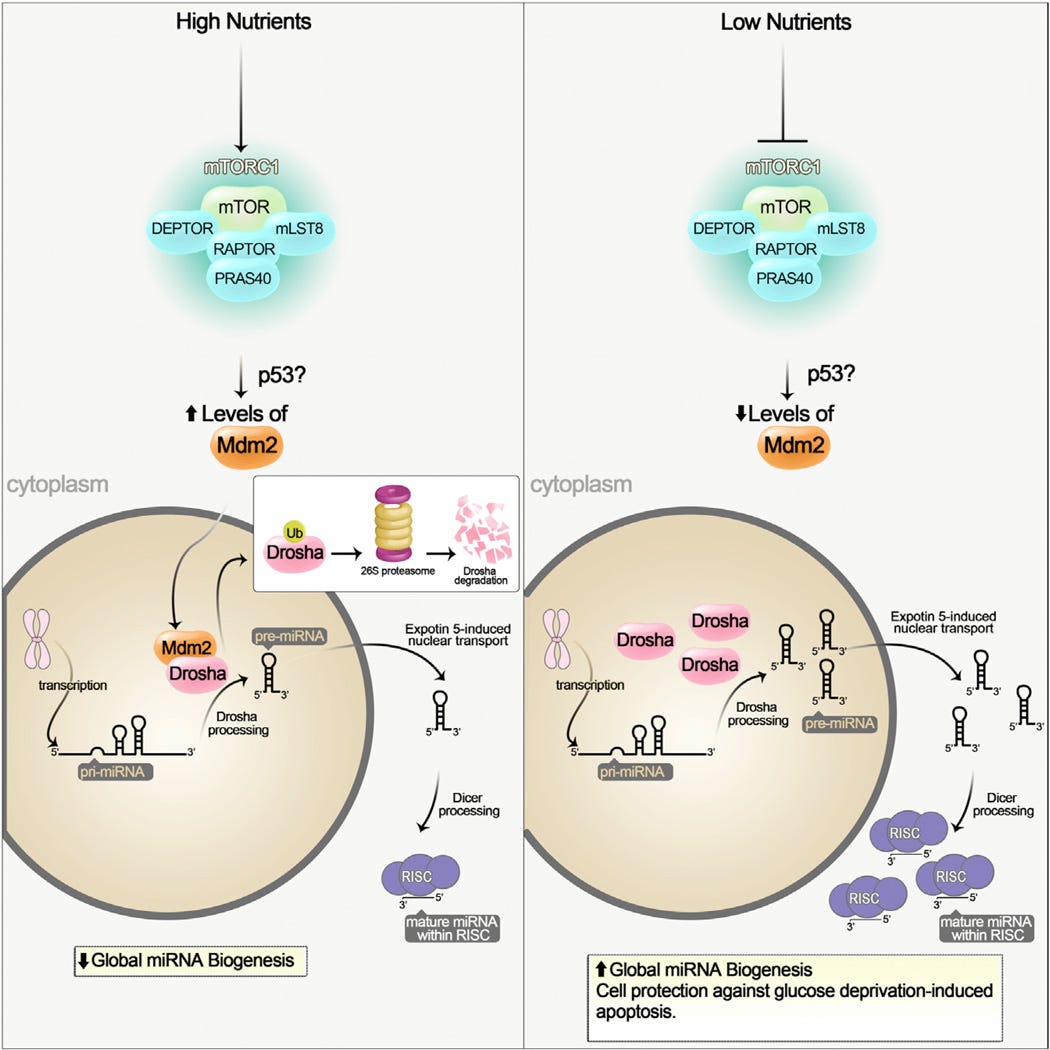
“In dieser Ausgabe von Molecular Cell zeigen Ye et al. (2015), dass mTORC1 die miRNA-Biogenese unter nährstoffreichen Bedingungen über die E3-Ubiquitin-Ligase Mdm2, die den Abbau von Drosha fördert, global reguliert.”
(…)
“miR-297, miR-376b-3p, miR-567 und miR-627-5p erhöhten die Resistenz der Drosha-silenced Zellen gegenüber Glukoseentzug. Zwei der vier miRNAs, miR-297 und miR-567, erhöhten den Drosha-Proteinspiegel signifikant, was darauf hindeutet, dass diese beiden miRNAs die Zellen direkt über den Drosha-Spiegel vor Apoptose schützen können. Somit scheint der mTORC1-Mdm2-Drosha-Weg eine wichtige Rolle bei der zellulären Anpassung an Glukoseentzug zu spielen.”
mTORC2 promotes cell survival through c-Myc–dependent up-regulation of E2F1

“Nach dem Ausschluss von miRNAs, die auf extrem niedrigem Niveau oder statistisch nicht signifikant (weniger als das Dreifache) exprimiert wurden, identifizierten wir 22 bzw. 20 unterschiedlich exprimierte miRNAs in Rapamycin- und PP242-behandelten Zellen im Vergleich zu Kontrollzellen (Abb. 1 B).”
(…)
“mTORC2, aber nicht mTORC1, reguliert miR-9-3p negativ und fördert das Zellüberleben
Um direkt zu bestätigen, ob mTORC2 den miR-9-3p-Spiegel beeinflusst, schalteten wir Rictor oder Raptor aus, indem wir siRNAs mit zwei unabhängigen Zielsequenzen verwendeten, die die Phosphorylierung von Akt (Ser 473) und S6 (Ser 235/236) hemmen (die wichtigsten Kennzeichen der mTORC1- bzw. mTORC2-Aktivierung; Abb. 2 A, rechts). Die Expression von reifem miR-9-3p wurde durch die Ausschaltung von Rictor, aber nicht von Raptor induziert, wie mittels RT-qPCR gezeigt wurde (Abb. 2 A, links). Darüber hinaus bestätigte die Northern-Blot-Analyse, dass sowohl der Vorläufer als auch das reife miR-9-3p durch die Hemmung von mTORC2, nicht aber von mTORC1 induziert werden (Abb. 2 B). Darüber hinaus war der miR-9-3p-Spiegel unter Serumentzug, der die mTORC1/2-Aktivität unterdrückt, erhöht (Abb. 2 C, links), während Aminosäurehunger, der spezifisch mTORC1 blockiert, nur geringe Auswirkungen auf miR-9-3p hatte (Abb. 2 C, rechts). Diese Ergebnisse zeigen eindeutig, dass miR-9-3p durch mTORC2, aber nicht durch mTORC1 negativ reguliert wird.”
mTORC2 promotes cell survival through c-Myc–dependent up-regulation of E2F1

“Die Inaktivierung von mTORC2 reduziert die PP2A-Aktivität gegenüber c-Myc-Serin 62 (S62), was zu einer verstärkten c-Myc-Phosphorylierung und -Expression und einer erhöhten Transkription von pri-miR-9-2/miR-9-3p führt, die wiederum E2F1 unterdrückt und die Apoptose fördert.”
(…)
”Eine erhöhte c-Myc-Aktivität induziert die Transkription von pri-miR-9-2/miR-9-3p, was wiederum die Expression von E2F1 hemmt, einem Transkriptionsfaktor, der für das Überleben von Krebszellen und die Tumorprogression entscheidend ist, was zu einer verstärkten Apoptose führt. In-vivo-Experimente mit B-Zell-spezifischen mTORC2-Deletionsmäusen (Rapamycin-unempfindlicher Begleiter von mTOR) und einem Xenotransplantat-Tumormodell bestätigten, dass die Inaktivierung von mTORC2 zu einer Hochregulierung von c-Myc und miR-9-3p, einer Herunterregulierung von E2F1 und folglich zu einer Verringerung des Zellüberlebens führt. Umgekehrt hob Antagomir-9-3p die durch mTORC1/2-Inhibitoren verstärkte E2F1-Unterdrückung und die daraus resultierende Apoptose in Xenotransplantaten auf. Unsere In-vitro- und In-vivo-Ergebnisse zeigen, dass mTORC2 das Zellüberleben fördert, indem es die E2F1-Expression durch einen c-Myc und miR-9-3p-abhängigenMechanismus stimuliert.”
Wie hier wunderschön erkennbar wird, haben wir bereits ein Huhn-Ei-Dilemma bei der Frage zu klären, ob miRNAs direkt via Signalmolekülinteraktion oder indirekt durch Transkriptionsfaktoren (C-Myc) nach der Signalkaskade angesteuert werden. Denn auch miRNAs werden im Nukleus rekrutiert und (teilweise) prozessiert.
MAPk
KRAS Hijacks the miRNA Regulatory Pathway in Cancer


“Interessanterweise deuten neuere Erkenntnisse auch darauf hin, dass onkogene Mutationen des KRAS (Kristen rat sarcoma viral oncogene homolog) zum Teil durch die Modulation der Aktivität von Mitgliedern des miRNA-Regelkreises wirken. Hier zeigen wir die entscheidende Rolle auf, die Mutationen in der miRNA-Kernmaschinerie bei der Förderung der malignen Transformation spielen. Darüber hinaus erörtern wir, wie mutiertes KRAS gleichzeitig mehrere Schritte der miRNA-Verarbeitung und -Funktion beeinflussen kann, um die Tumorentstehung zu fördern. Obwohl die Fähigkeit von KRAS, den miRNA-Regulationsweg zu unterwandern, seine onkogene Natur noch komplexer macht, bietet sie auch einen potenziellen therapeutischen Weg, der in der Klinik noch nicht genutzt wurde. Darüber hinaus stellt die gleichzeitige Beeinflussung von mutiertem KRAS und Mitgliedern der miRNA-Kernmaschinerie eine potenzielle Strategie zur Behandlung von Krebs dar.”
KRAS regulation of miRNA: Stepping on the brake to go faster

“KRAS stimuliert die Hyperaktivierung von nachgeschalteten Effektor-Signalnetzwerken (z. B. RAF-MEK-ERK-MAPK), die globale Veränderungen im Transkriptom und Proteom bewirken. Diese globalen Veränderungen können auf transkriptioneller, posttranskriptioneller, translationaler und posttranslationaler Ebene reguliert werden.
Eine Klasse von posttranskriptionellen Elementen, die weitreichende Veränderungen in der Proteomlandschaft bewirken, sind microRNAs (miRNAs). miRNAs sind kurze, nicht codierende Nukleotide (~21 nt), die durch Bindung an Proteine der Argonaute-Familie (AGO1-4), vor allem AGO23, eine Vielzahl von RNA-Zielen regulieren. Die miRNA-AGO2-Komplexe erkennen komplementäre RNA-Sequenzen (2-7 nt), was zu deren Abbau durch den RNA-induzierten Silencing-Komplex (RISC) führt. Kürzlich entwickelten Li und Kollegen eine In-vivo-Strategie zur Isolierung von AGO2-miRNA-RNA-Komplexen in Mäusegeweben mit Hilfe einer Halo-Enhanced Ago2 Pull-down (HEAP) Affinitätsreinigungsmethode, die eine systemweite Bestimmung von miRNA-Targets ermöglicht. Shui et al. nutzen nun diesen HEAP-Assay, um die Aktivität von miRNAs in genau definierten, genetisch veränderten Mausmodellen mit Expression von KrasWT oder KrasG12D im Kolonepithel und in Kolontumoren zu bewerten. Sie nutzten ihre transkriptomischen, proteomischen und phosphoproteomischen Datensätze in diesen Mausmodellen, um einen komplexen Mechanismus der KrasG12D-abhängigen miRNA-Funktion aufzudecken.”
Kurze Anmerkung:
WT = Wild Type
G12D = Mutiert
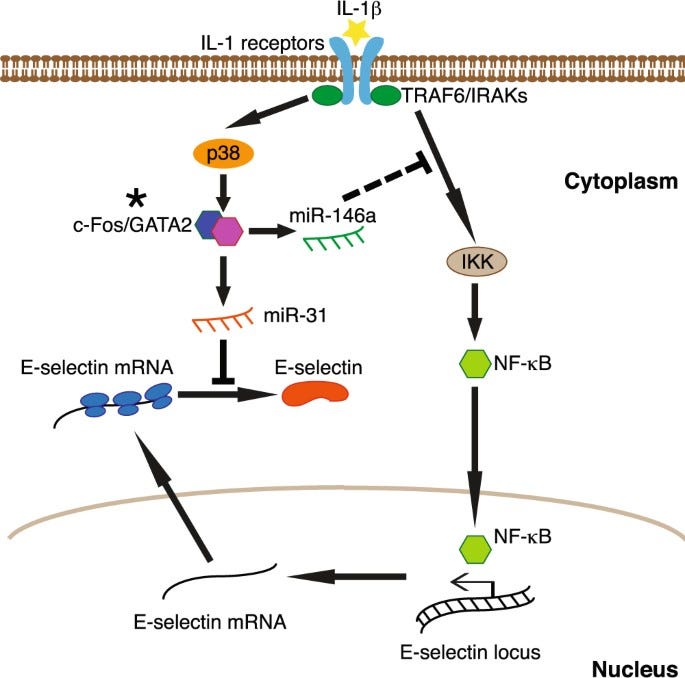
“Als Reaktion auf IL-1β unterdrückt die p38 MAP-Kinase die Expression von E-Selektin auf transkriptioneller und posttranskriptioneller Ebene über miR-146a bzw. miR-31. Diese Ergebnisse zeigen neue Mechanismen auf, durch die p38 die Expression von E-Selektin durch verschiedene microRNAs nach entzündlichen Stimuli, die mit dem Fortschreiten von Krebs verbunden sind, herunterreguliert.”

“JNK1 und c-Jun erwiesen sich als wichtige Ziele von DCA, die vor p53 liegen und den miR-34a-Signalweg und die Apoptose in Gang setzen. Schließlich wurde gezeigt, dass die Aktivierung dieses proapoptotischen JNK1/miR-34a-Kreislaufs auch in vivo in der Rattenleber stattfindet.”
cAMP
A cAMP-response element binding protein-induced microRNA regulates neuronal morphogenesis
“Obwohl die räumliche und zeitliche Strukturierung der miRNA-Expression streng kontrolliert wird, ist wenig über die Signale, die ihre Expression auslösen, und die Mechanismen ihrer Transkriptionsregulierung bekannt. Darüber hinaus sind nur wenige miRNA-Targets experimentell validiert worden. Die miRNA miR-132 wurde in einem genomweiten Screening als Ziel des Transkriptionsfaktors cAMP-response element binding protein (CREB) identifiziert.”
Wir braten heute CREBs. Mhhhh lecker!

“Faktoren, die den cAMP-Spiegel erhöhen, können die linienspezifische Differenzierung von Gliomzellen in astrozytenähnliche Zellen auslösen. Das Differenzierungsmuster und die zugrunde liegenden Mechanismen sind jedoch nach wie vor unklar. Wir haben herausgefunden, dass die durch cAMP/Proteinkinase A (PKA)/cAMP responsive element binding protein 1 (CREB1) induzierte miR-221/222-Unterdrückung zur neuronenartigen Differenzierung von Gliomen beiträgt.”
Regulation of the miR-212/132 locus by MSK1 and CREB in response to neurotrophins
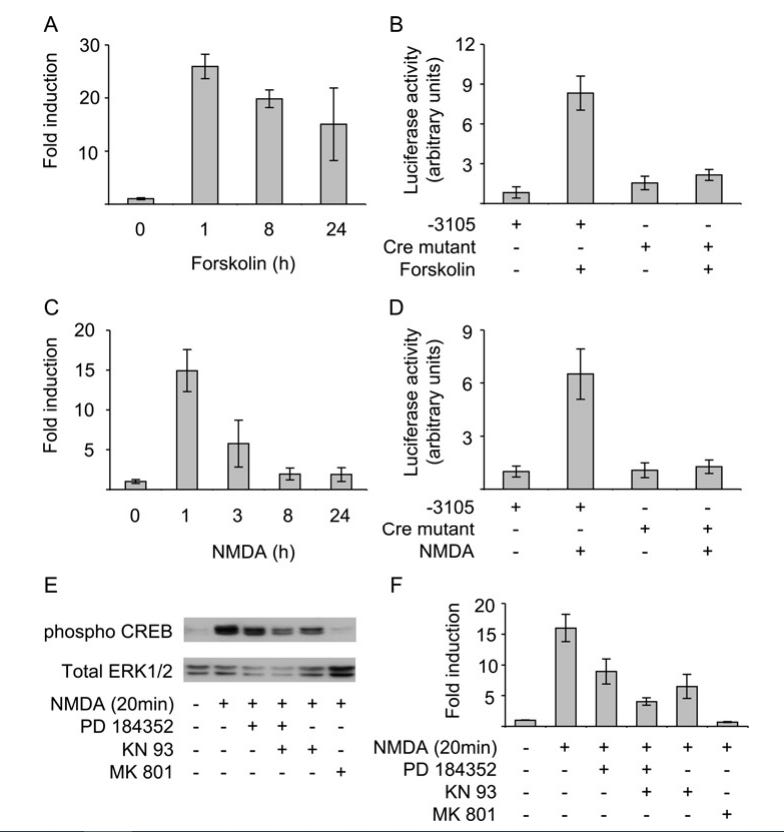
“Forskolin erhöht den cAMP-Spiegel, was zu einer PKA-Aktivierung (Proteinkinase A) und CREB-Phosphorylierung in einer Reihe von Zelltypen, einschließlich kortikaler Neuronen, unabhängig von ERK1/2 und MSK1/2 führt. Die Stimulation mit Forskolin führte zu einem anhaltenden Anstieg von pri-miR-212/132 in kortikalen Neuronen (Abbildung 7A). Im Einklang damit konnte Forskolin die Induktion des miR-212/132-Promotorkonstrukts stimulieren. Die Mutation der vier Cre-Stellen in diesem Konstrukt blockierte die Induktion (Abbildung 7B).”
Regulation of microRNA-375 by cAMP in Pancreatic β-Cells
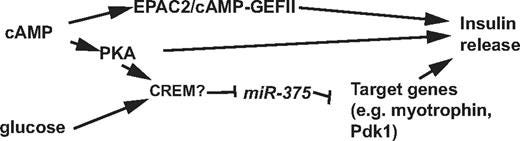
“Wie bereits berichtet, reguliert miR-375 die Insulinsekretion negativ, und eine Abschwächung von miR-375 über den cAMP-PKA-Weg könnte die Insulinantwort in β-Zellen der Bauchspeicheldrüse fördern.”
Und wer bis hier hin denkt, das sei schon schweinekompliziert, der möge sich mal folgenden Witz, was “Crosstalks” angeht, auf der Zunge zergehen lassen:
The interplay between transcription factors and microRNAs in genome-scale regulatory networks
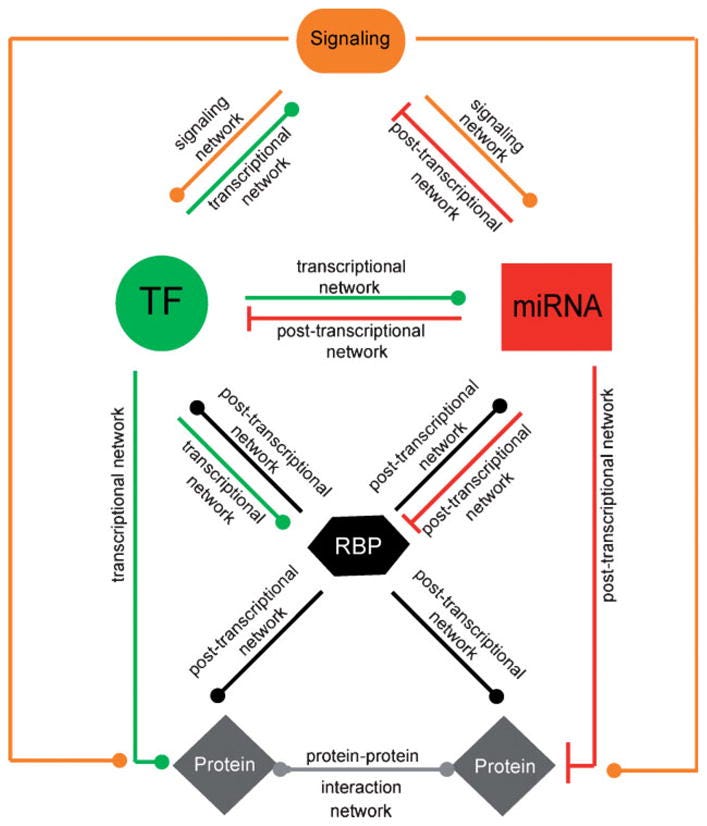
“Bei der Untersuchung des Grades der TFs und miRNAs, die an Rückkopplungsschleifen im integrierten C. elegans-Netzwerk beteiligt sind, stellten wir fest, dass die meisten Teilnehmer der Schleife sowohl einen hohen in- als auch out-Grad aufweisen. In den meisten miRNA ↔TF Rückkopplungsschleifen regulieren die miRNAs viele TFs und werden von vielen TFs reguliert und umgekehrt. Die Tatsache, dass sowohl die miRNAs als auch die TFs, die an Rückkopplungsschleifen beteiligt sind, viele nachgeschaltete Ziele haben, bedeutet, dass nicht nur die Expression der TF und der miRNA, sondern auch die ihrer jeweiligen Ziele eng koordiniert ist. Auf der Grundlage dieser Beobachtungen schlagen wir vor, dass sich die von TFs und miRNAs ausgeübten genregulatorischen Wirkungen auf große Mengen von Genen „ausbreiten“ können. Diese „regulatorische Ausbreitung“ könnte prinzipiell für die Regulierung großer Mengen von Genen oder Genbatterien wichtig sein, z. B. in verschiedenen Geweben oder als Reaktion auf Entwicklungs- oder Umweltfaktoren (Abb. 3A). Durch die Integration menschlicher Protein-Protein-Interaktionen mit miRNA-Target-Interaktionen fanden Liang und Li heraus, dass Proteine, die von mehreren miRNAs reguliert werden, tendenziell einen höheren Grad an Konnektivität in Protein-Protein-Interaktionsnetzwerken aufweisen. Das Targeting von Protein-Protein-Interaktions-Hubs durch viele miRNAs kann folglich auch eine große Anzahl von interagierenden Proteinen beeinflussen (Abb. 3B).”
Und immer daran denken: miRNAs werden auch noch von lncRNAs (long non coding RNAs) reguliert während lncRNAs sich auch noch gegenseitig regulieren.
Mechanisms of circRNA/lncRNA-miRNA interactions and applications in disease and drug research


Uff… Soweit so schick also.
Dann gucken wir uns jetzt die Transkriptionsfaktoren kurz an? Da hier so viel umfangreiche Lektüre existiert und jede einzelne Signalkaskade so extrem viele ansteuert und dahingehend auch sehr viel kontroverse Dinge beobachtet werden, halte ich diesen Teil ganz allgemein. Ich möchte mich auf eine spezifische Transkriptionsfaktorenfamilie fokussieren, die ich in meinem letzten Teil auch noch einmal erwähnen werde. Zur kurzen Klärung was Transkriptionsfaktoren sind:

“Transkriptionsfaktoren, kurz TF, sind regulatorische Proteine, die durch Bindung an spezifische Regionen in der DNA die Rekrutierung der RNA-Polymerase und den Start der Transkription positiv oder negativ modulieren.
2. Struktur
Trotz der unterschiedlichen Gestaltung verschiedener Transkriptionsfaktoren, lässt sich ihre Struktur generalisieren. Die aktive Form ist oftmals ein homo- oder heterodimer, die folgende Domänen enthalten:
DNA-bindende Domäne (DBD)
Die DBD vermittelt die Bindung an die DNA. Sie enthält positiv geladene Aminosäuren, die mit der negativ geladenen DNA interagieren können. Die Struktur dieser der DBD kann jedoch unterschiedlich sein und besteht aus mehreren typischen Motiven:
Leucin-Zipper
Helix-Turn-Helix
Helix-Loop-Helix
Zinkfinger
Transkriptionsaktivierende Domäne (TAD)
Die Domäne bindet weitere regulatorische Aktivatoren.
Zusätzliche Domänen:
Dimerisierungsdomäne
Inihbierende Domäne
Aktivierende Domäne”
Auf in die Vollen!
E2F-Transkriptionsfamilie

“Darüber hinaus haben wir PKC delta und eta als die Isoformen identifiziert, die speziell an der Induktion des proteasomalen Abbaus von E2F1 beteiligt sind. Wir zeigen auch, dass die Herunterregulierung von E2F1 durch neue PKC-Isoenzyme die Aktivierung der mitogen-aktivierten Proteinkinase (MAPK) p38beta erfordert.”
“Die E2F-Transkriptionsfaktoren sind grundlegende Regulatoren der Zellzyklusprogression, der Differenzierung, der DNA-Reparatur und der Apoptose.”
Cell cycle oscillators underlying orderly proteolysis of E2F8
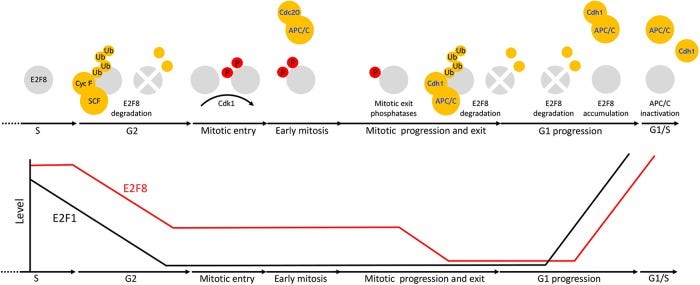
“E2F8 ist ein Transkriptionsrepressor, der E2F1 an der Schnittstelle zwischen Zellzyklus, Apoptose und Krebs antagonisiert. Zuvor hatten wir entdeckt, dass E2F8 ein direktes Ziel der APC/C-Ubiquitin-Ligase ist. Dennoch bleibt unbekannt, wie E2F8 während des gesamten Zellzyklus dynamisch kontrolliert wird. Mit Hilfe neu entwickelter humaner zellfreier Systeme, die verschiedene intermitotische und G1-Phasen sowie einen kontinuierlichen Übergang von der Prometaphase zu G1 nachbilden, konnten wir einen ineinandergreifenden Dephosphorylierungsschalter nachweisen, der den Abbau von E2F8 mit dem mitotischen Exit und der Aktivierung von APC/CCdh1 koordiniert. Darüber hinaus decken wir unterschiedliche Proteolyseraten für E2F8 zu verschiedenen Zeitpunkten innerhalb der G1-Phase auf, was die Akkumulation von E2F8 im späten G1 erklärt, während APC/CCdh1 noch aktiv ist. Schließlich zeigen wir, dass das F-Box-Protein Cyclin F E2F8 in der G2-Phase reguliert. Insgesamt definieren unsere Daten die Regulierung von E2F8 während des gesamten Zellzyklus und beleuchten eine umfassende Koordination zwischen Phosphorylierung, Ubiquitinierung und Transkription im Säugetierzellzyklus.”
DNA‐damage response control of E2F7 and E2F8

“Diese Ergebnisse deuten darauf hin, dass E2F7 und E2F8 eine wichtige Rolle bei der Regulierung der Aktivität von E2F1 während der DNA-Schädigung spielen. Da E2F7 und E2F8 als Komplex am E2F1-Promotor existieren, wird dies wahrscheinlich durch die Fähigkeit von E2F7 und E2F8 vermittelt, die E2F1-Transkription zu unterdrücken”
Gut, gut, nachdem ich mich jetzt also mühsam downstream durchgeackert habe und alle Aspekte meiner Folie im Detail diskutierte, freue ich mich tatsächlich darauf, im kommenden Substack all das, was ich hier herausarbeitete, auf diese Genplörren runter zu brechen und euch ein paar unschöne Fragen zu stellen, die weder Pfizer, noch BioNTech, noch Moderna jemals beantworteten.
Ich entlasse euch heute mal mit einem megafiesen Cliffhanger und einer Anspielung darauf, wieso mir die E2F-Familie so wichtig erscheint.
. Bottom, Model with biphasic contraction phase (i.e., Δ > 0 and α > 0). Insets, The same data for the entire time series of 921 days. See Table I for parameter estimates.")
“Für CD8+ und CD4+ T-Zellen beträgt die Verdopplungszeit während der anfänglichen Expansionsphase 8 bzw. 11 Stunden. Die Halbwertszeit während der Kontraktionsphase nach dem Höhepunkt der Reaktion beträgt 41 Stunden bzw. 3 Tage.”
Measurement of macrophage growth and differentiation

“Je nach ihrem Differenzierungsgrad wachsen Makrophagenpopulationen unterschiedlich schnell. So haben beispielsweise primäre Makrophagen aus dem Knochenmark (BMM) eine durchschnittliche Verdopplungszeit von 20 Stunden, während aus Splenozyten gewonnene Makrophagen eine Verdopplungszeit von ~30 Stunden haben (Chitu et al., 2009).”
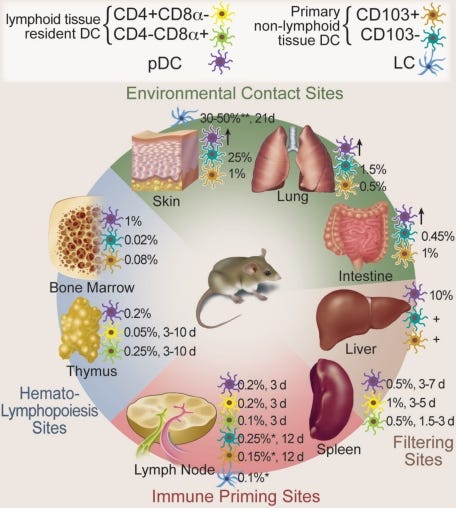
“DCs sind im ganzen Körper verteilt und in lymphatischen Organen und an Kontaktstellen mit der Umwelt angereichert. Die Halbwertszeit von DCs im stationären Zustand beträgt Tage bis hin zu einigen Wochen, und sie müssen durch proliferierende hämatopoetische Vorläuferzellen, Monozyten oder gewebeeigene Zellen ersetzt werden.”
Hm. Liest sich das danach an, dass man in peripheren Immunzellen aus einer Blutprobe E2F8 aber nicht E2F1 in einer log2fold GSEA-Analyse um das 1.5 fache überexpressiert messen sollte und dann von einer adäquaten Immunantwort schwafeln kann?
In diesem Sinne: Just say narf und ich freue mich auf den letzten Teil.