Downstream Teil 4
Meine erste Folie, mit der ich zufrieden bin und die mir zeitgleich Angst macht.

4. Das Huhn-Ei Problem der Crosstalks: Was war zuerst da?: c(yclic)AMP, MAPk, JAK/STAT oder mTORC1/2?

Mit der Antwort “so funktioniert es nicht” kann man bei dieser Frage nichts verkehrt machen.
Was verstehen wir denn überhaupt unter Crosstalks?
Vllt. ist es mit einem Beispiel am Einfachsten zu verstehen:
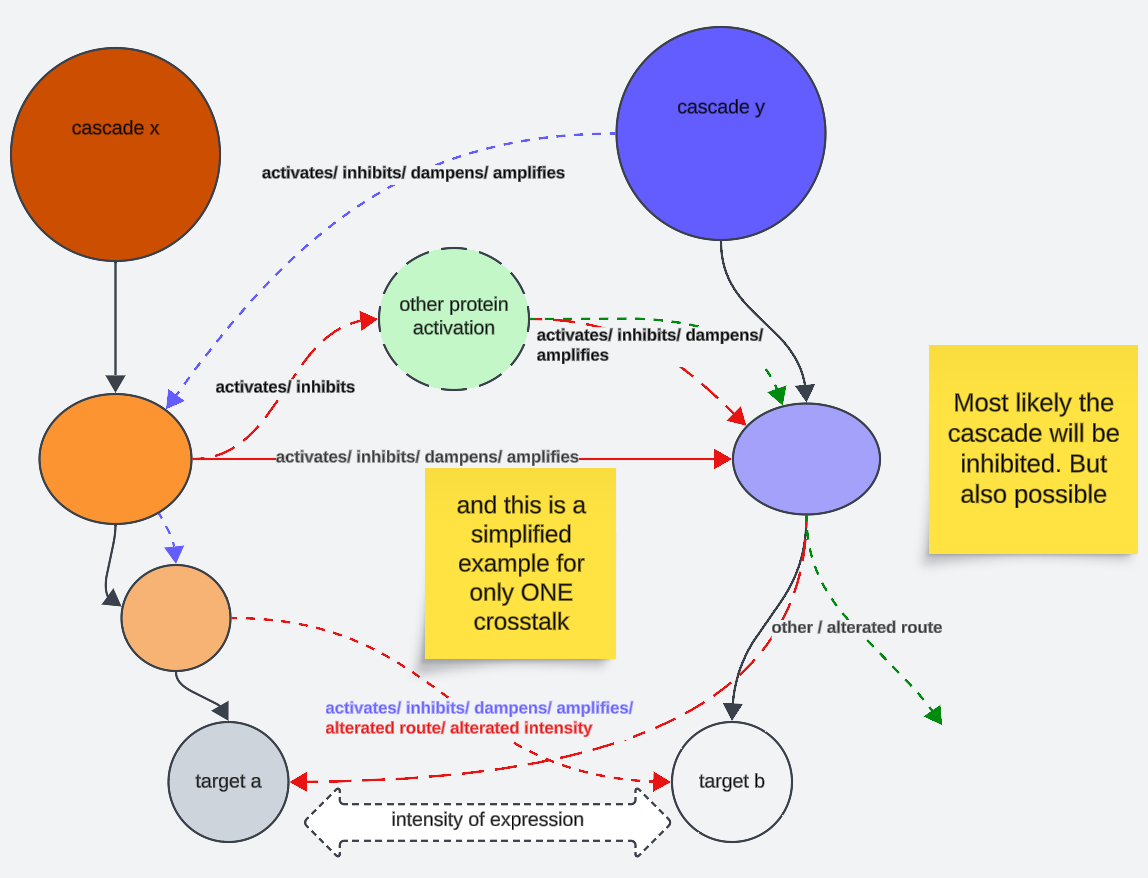
Wir folgen [Kaskade Y]. Der “traditionelle”/kanonische Weg wären die fetten schwarzen Pfeile. Nun beeinflusst jedoch [Kaskade X] [Kaskade Y] in ihren spezifischen downstream Molekülinteraktionen. Sie könnte also die [Kaskade Y] a) inhibieren b) verstärken c) überhaupt den weiteren Fluss downstream aktivieren, ohne das die erste Molekülreaktion im traditionellen Weg downstream stattfindet d) den Expressionsweg der weiteren Kaskade alterieren und ein anderes Ziel dadurch binden f) zu einem weiteren Crosstalk mit [Kaskade X] führen, der dann [Kaskade X] wiederum ähnlich affektiert. Eine weitere Möglichkeit wäre das gegenseitige Beeinflussen der Ziele a und b, was beispielsweise die Expressionsrate und Expressionsdauer angeht.
In diesem Passus werde ich mich - allerdings nur kurz anreißend, da es sonst echt ausufern würde - also mit den Crosstalks zwischen 4 der größten und garantiert affektierten (grüne Pfeile) Signalkaskaden beschäftigen. Und für viele Crosstalks, die ich noch nicht anschnitt, ein paar Beispiele liefern.
Doch bevor ich das kann, müssen wir fix klären, warum das Timing die Interaktionen maßgeblich beeinflussen wird:
Zellsignale, die so ziemlich jeden Aspekt der Metabolismen regulieren, laufen also zeitgleich ab. Darum gibt es auch keine Antwort auf die Frage, “mit welcher Kaskade” es denn anfängt, da es - wie bereits erwähnt - absolut von Zeit, Raum, Distanz, Kontext und Intensität des eingehenden Signals/ Proteins/ Hormons/ Zytokins/ Ions/ etc. abhängig wäre, genauso wie von dem Zustand der jeweiligen Zelle, welche Crosstalks mit welchen Bedingungen gerade getriggert werden.
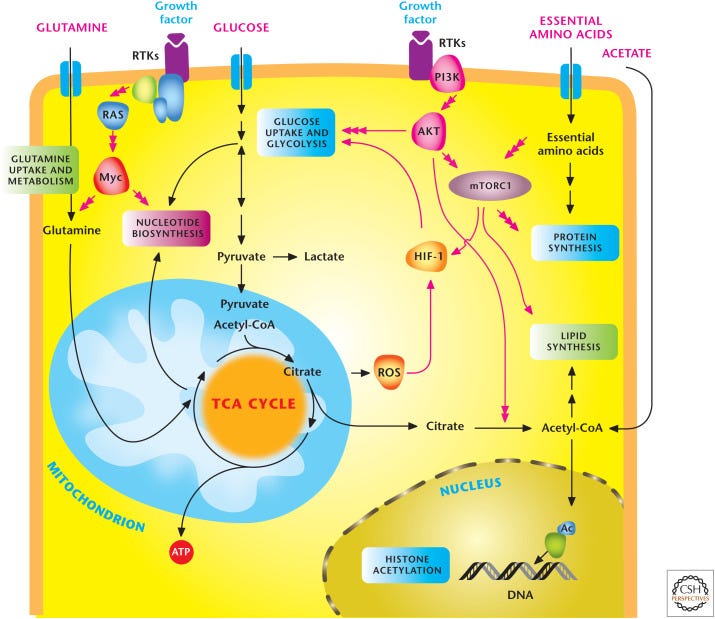
Noch dazu haben wir ein riesen Problem, welches erstmals 2023 wirklich detailliert diskutiert wurde: Nichtlineare Signaltransduktion!

“Eine Ratenschwelle ist definiert als ein Schwellenwert in der Änderungsrate der Umgebung, bei dem ein Ratenwert unterhalb der Schwelle keine Signalisierung aktiviert und ein Ratenwert oberhalb der Schwelle zu einer Signalaktivierung führt. Wir haben die p38/Hog1-Signalgebung bei osmotischem Stress in Hefe, Chemotaxis und Stressreaktion in Bakterien, zyklische Adenosinmonophosphat-Signalgebung in Amöben, Wachstumsfaktor-Signalgebung in Säugetierzellen, Morphogen-Dynamik während der Entwicklung, zeitliche Dynamik der Glukose- und Insulin-Signalgebung und räumlich-temprorale Stressoren in der Niere untersucht. Diese Beispiele aus der Literatur zeigen, dass Schwellenwerte weit verbreitet sind und eine unterschätzte grundlegende Eigenschaft der Zellsignalgebung darstellen. “
(…)
“Im Gegensatz dazu erfahren Zellen in jedem Organismus oder in jeder natürlichen Umgebung eine Vielzahl von allmählichen Veränderungen der äußeren Reize. Die derzeitigen Behandlungen und Therapien basieren auf beobachteten Phänotypen (violette Ebene) und aktuellen Krankheitsmodellen (grauer Stern) bei solchen akuten Behandlungen, wobei die Reaktion einer Zelle auf den Stimulus unter physiologischen Bedingungen möglicherweise übersehen oder falsch interpretiert wird. Nimmt man beispielsweise ein Dutzend Konzentrationen aus dem oben erwähnten Dosis-Wirkungs-Experiment und ändert die Konzentration akut alle 1 Minute für 30 Minuten, dann sind 12^30 = 2,3*10^32 einzigartige Kombinationen möglich. Dieses einfache Beispiel zeigt, dass es unendlich viele Möglichkeiten gibt, die Umgebung von Zellen auf nicht-akute Weise zu verändern. Diese neue Dimension der dynamischen Veränderungen des Stimulusmoleküls kann unseren phänotypischen Raum, der derzeit im Labor nicht erforscht wird, dramatisch erweitern (weißer Stern). Die Einbeziehung dieser kritischen Dimension wird es uns ermöglichen, physiologisch relevante Phänotypen bei verschiedenen dynamischen Phänotypen (gelbe Ebene) und vor verschiedenen genetischen Hintergründen (cyanfarbene Ebene) zu beobachten.”

“Im Vergleich dazu führen dieselben Zellen und Signalwege, die einer quadratisch ansteigenden Bedingung ausgesetzt sind (Abbildungen 11G-I), zu einer Verzögerung der Signalaktivierung (Q1), zur Erkennung einer Konzentrationsaktivierung (Q2) und von Aktivierungsschwellen für die Rate (Q3), zu einer linearen Konzentrations- (Q4) und einer linearen Ratenabhängigkeit (Q5) sowie zu einer Sättigungskonzentration und einer Ratenschwelle (Q6), nach der die Signalisierung nicht mehr ratenabhängig ist, und eine Erhöhung des Stimulussignals führt nicht zu einer Erhöhung der Signalisierung. Dieses Beispiel veranschaulicht, dass nicht-akute Bedingungen entscheidend sein können, um verborgene Signalisierungsmerkmale zu entschlüsseln, die mit den derzeitigen akuten Störungsparadigmen nicht beobachtet werden können.”
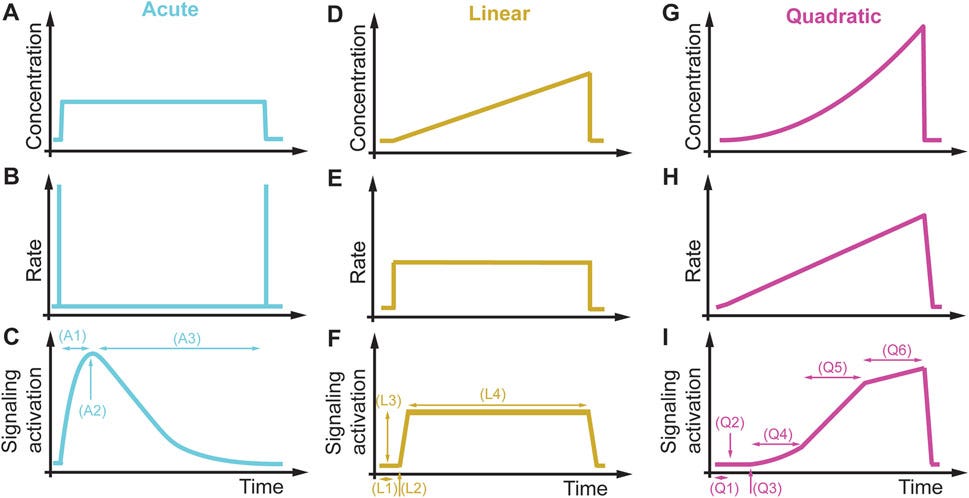
Hört noch jemand gerade schleifende Zahnräder und eine Uhr ticken?

“Darüber hinaus haben „redundante Phosphatasen“ spezifische, nicht-redundante Funktionen, die nur unter nicht-akuten Umweltbedingungen nachweisbar sind. Wir spekulieren, dass ein mögliches Netzwerkmotiv, das zu einer solchen Ratenschwelle in einem sich anpassenden System führen könnte, eine Kombination aus einer Hill-Funktion mit einer negativen Rückkopplungsschleife oder mit einer inkohärenten Vorwärtsschleife ist (Milo et al., 2002; Ma et al., 2009; Rahi et al., 2017). Im Kontext der Signaltransduktion beschreibt eine Hill-Funktion die Beziehung zwischen der Konzentration eines Liganden und der nichtlinearen Aktivierung eines nachgeschalteten Signalproteins. Eine negative Rückkopplungsschleife ist die Regulierung eines vorgeschalteten Signalproteins durch ein nachgeschaltetes Signalprotein. Eine inkohärente Feed-Forward-Schleife hat die Eigenschaft, sowohl ein Ziel-Signalprotein als auch einen Inhibitor dieses Proteins zu aktivieren, der dann das Ziel-Signalprotein hemmt. In dieser Studie wurde die Hypothese aufgestellt, dass der neuartige Mechanismus der Ratenschwelle in der Zellsignalgebung auch in anderen Signalwegen und Organismen vorkommen könnte, was im Mittelpunkt dieses Übersichtsartikels steht.”
So. Nachdem wir also das Regelwerk definiert haben, äh, oder eben auch nicht, weil dieses komplexe Netzwerk aus Signalmolekülen nicht mal ansatzweise begriffen ist, können wir ja endlich mit den Crosstalks durchstarten. Juchhu!
cAMP (cyclic Adenosine MonoPhosphate)
Inhibition of cytokines and JAK-STAT activation by distinct signaling pathways.

„Wir beschreiben die Unterbrechung der durch Zytokine ausgelösten JAK-STAT-Signale durch cAMP, das Kalziumionophor Ionomycin und den Granulozyten/Makrophagen-Kolonie-stimulierenden Faktor.”
Activation of Protein Kinase A Inhibits Interferon Induction of the Jak/Stat Pathway in U266 Cells (Und ja: Das Paper ist bis heute gerne zitiert und relevant)

“Die Hemmung der IFNβ-stimulierten GRR-Bindung korrelierte mit dem dosisabhängigen Forskolin-induzierten Anstieg des intrazellulären cAMP, was bestätigt, dass der Anstieg des cAMP in diesen Zellen die IFNβ-Aktivierung des Jak/Stat-Signalwegs hemmen könnte.”

“Neben der direkten Regulierung vieler wichtiger zellulärer Prozesse beeinflusst cAMP eine Reihe von intrazellulären Signalwegen, wie z. B. die MAP-Kinase-Wege, die zyklinabhängige Kinase, die Ca2+-abhängigen Signalwege und den Jak/STAT-Weg. Jüngste Studien weisen darauf hin, dass die cAMP-abhängige Signalübertragung auch eng mit dem PI3K/PKB-Signalweg verwoben ist. Der PI3K/PKB-Signalweg ist eine Schlüsselkomponente bei der Kontrolle des Überlebens und der Vermehrung von Zellen. cAMP wird mit der Modulation der PKB-Aktivität in Verbindung gebracht; eine Erhöhung der intrazellulären cAMP-Konzentration kann jedoch entweder zu hemmenden oder stimulierenden Effekten auf die PKB-Aktivität führen. Die Rolle, die Epac in diesem Prozess spielt, ist derzeit noch unklar.
Da cAMP die PKB-Aktivität entweder hemmen oder stimulieren kann, ist es denkbar, dass Epac und PKA, die als nachgeschaltete Rezeptoren fungieren, diese Wirkungen unterschiedlich vermitteln, z. B. hemmt PKA die PKB-Aktivierung, während Epac/cAMP-GEF als positiver Modulator von PKB als Reaktion auf cAMP wirken könnte.”
Eigentlich zeigt dieses Paper schon ziemlich perfekt sämtliche cAMP Crosstalks, die ich in meiner Folie einzeichnete. Was sich wohl leicht erklären lässt, schaut man sich diese hübsche Folie dazu an:
https://www.cusabio.com/pathway/cAMP-signaling-pathway.html

Regulation of the JAK2-STAT5 Pathway by Signaling Molecules in the Mammary Gland
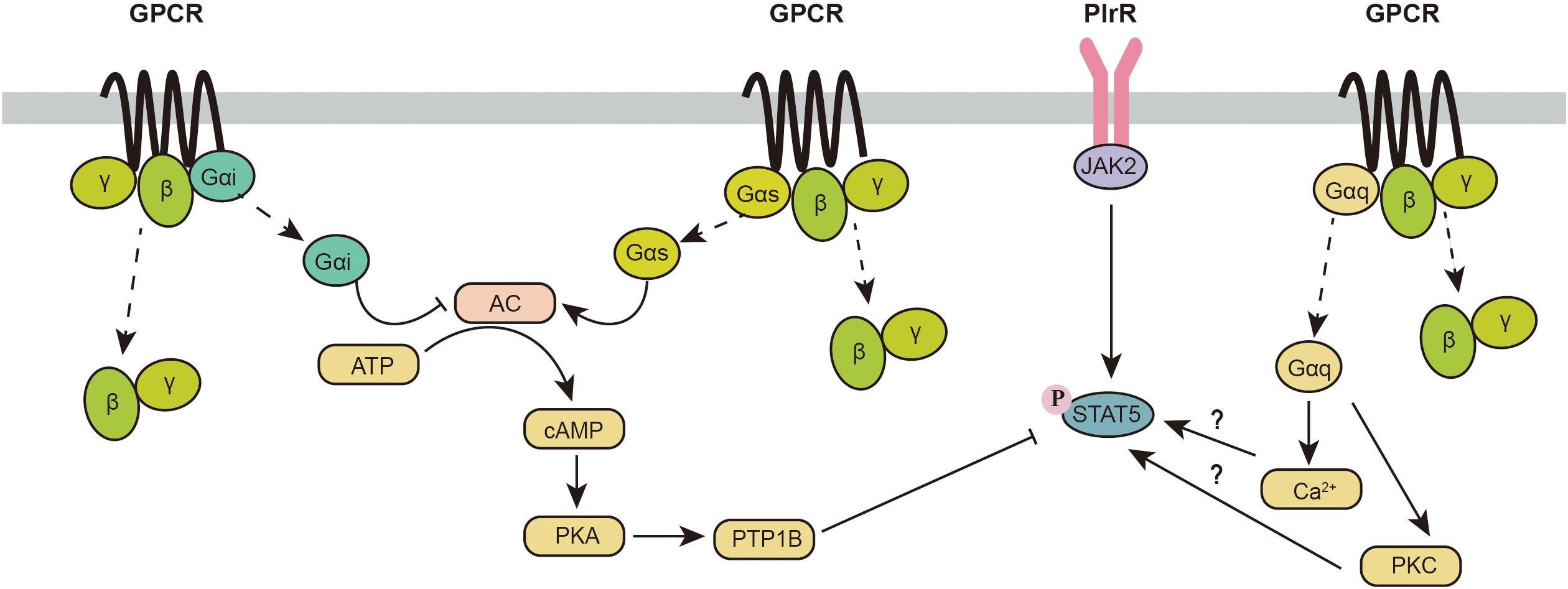
Ich denke das reicht, um zu zeigen, wie cAMP schon mal direkt die STATs affektiert. Beispiel: Aktivierung PKA → Aktivierung PTP1B → Inhibierung STAT5. Was hierbei hoffentlich jedem bewusst wird: Es ist ein extremes Check und Balance-Netzwerk welches darauf aufbaut, möglichst jeden Prozessschritt, der in Protein-Protein-Interaktionen stattfindet, extrem engmaschig mit jeder Menge “Notreißleinen” kontrollieren zu können.
Für die MAPk-Crosstalks packe ich einfach mal ein paar hübsche Folien nach, die selbsterklärend sein dürften. (Verwirrung: mein Hase, die mir bei meinen Rechtschreib, äh, Problemchen hilft, lacht gerade laut? Meint, dass jeder an dieser Stelle lachen wird? Wie meint sie das? 😒)
+
https://pmc.ncbi.nlm.nih.gov/articles/PMC203360/figure/cdd293f9/
(Leider ist diese Grafik in der endgültigen Publikation nicht mehr drin)

Contributions of extracellular-signal regulated kinase 1/2 activity to the memory trace
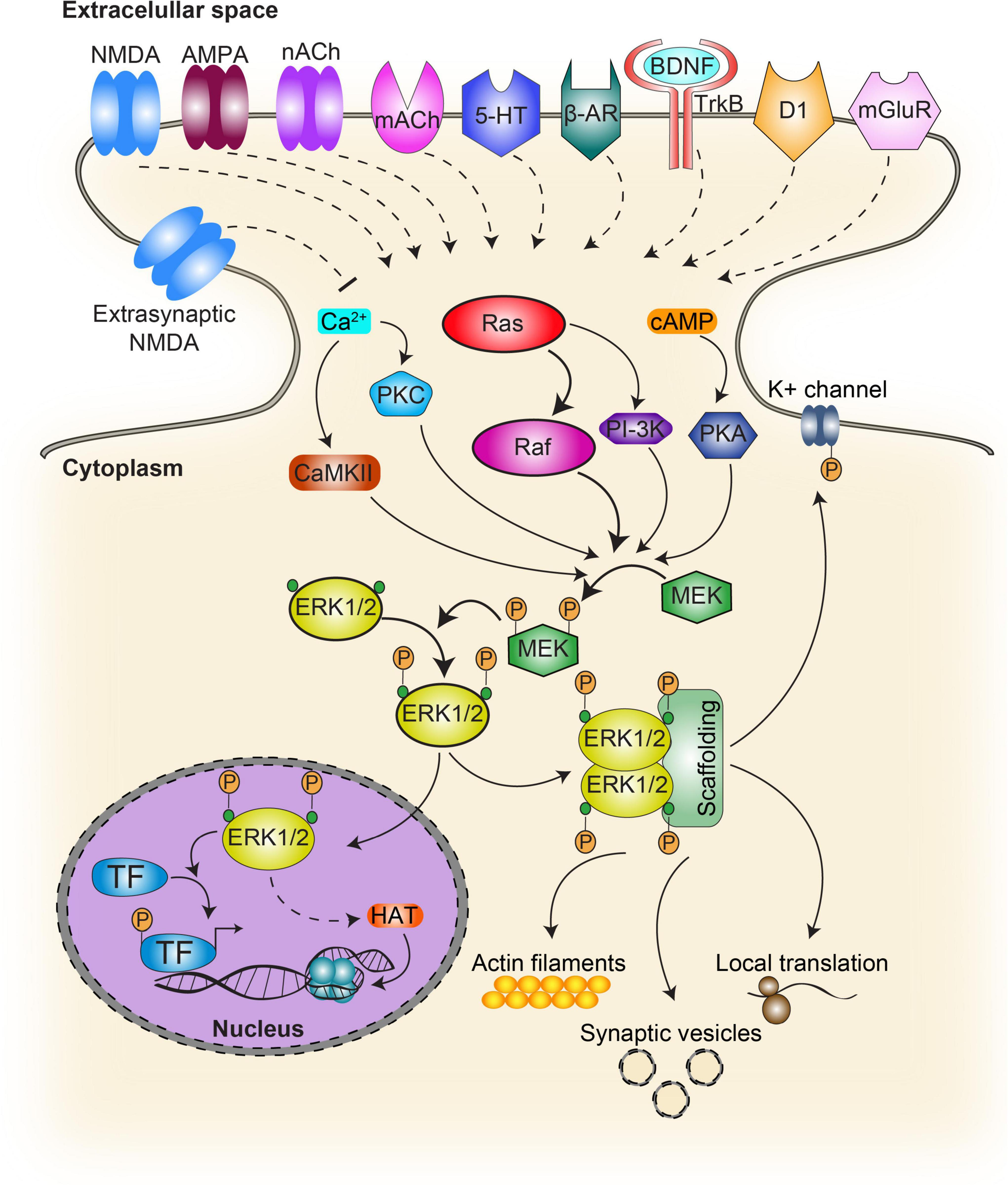
MAPkinasen sind keine Einbahnstraße und eines der größten Netzwerke überhaupt.😉

“Durch die Phosphorylierung des aktivierenden Transkriptionsfaktors 2 (ATF-2) und des Peroxisom-Proliferator-aktivierten Rezeptors gamma (PPARgamma) Coativator 1alpha (PGC-1alpha), Mitglieder zweier unterschiedlicher Kernfaktor-Familien, kontrolliert p38 MAPK die Expression des UCP1-Gens durch ihre jeweiligen Interaktionen mit einem cAMP-Reaktionselement und einem PPAR-Reaktionselement, die sich beide innerhalb eines kritischen Enhancer-Motivs des UCP1-Gens befinden.”
Quizfrage an dieser Stelle: Wie war das gleich wieder mit den Menstruationsstörungen? → Beschäftigt euch mal ein wenig mit Coativatoren.🤐
Und noch als Leseempfehlung:
Crosstalk between cAMP and MAPkinase signaling in the regulation of cell proliferation

Auf, auf zum mTORC!

“Es gibt immer mehr Belege dafür, dass mTORC2 durch GPCR-Signalgebung aktiviert wird. β- und α-Adrenozeptoren erhöhen den cAMP-Spiegel und fördern die Glukoseaufnahme über mTORC2, sind aber unabhängig von der PI3K/Akt-Signalisierung.”
Cyclic AMP Inhibits Akt Activity by Blocking the Membrane Localization of PDK1

“cAMP hemmt die Kinase-Aktivität von Akt in Vivo
Um zu verstehen, wie der cAMP-abhängige Zellsignalweg Akt moduliert, untersuchten wir die Auswirkungen von Forskolin, einem Aktivator der Adenylylzyklase, auf die Proteinkinase-Aktivitäten von Akt, die durch verschiedene Agonisten für die Zellproliferation wie EGF, Phorbol 12-Myristat 13-Acetat, Calyculin A und Serum induziert werden. Wir stimulierten pCMV6-HA-Akt-transfizierte COS-Zellen mit verschiedenen Agonisten und behandelten sie anschließend mit Forskolin (siehe Abb.1 A). Die Proteinkinase-Aktivitäten von Akt wurden durch EGF und Calyculin A stark und durch Serum schwach stimuliert, wie bereits berichtet. Interessanterweise wurden die von all diesen Agonisten induzierten Akt-Aktivitäten durch die Behandlung mit Forskolin stark gehemmt (Abb. 1 A). Daher beschlossen wir, weiter zu untersuchen, inwieweit die cAMP-abhängige Signalübertragung an der Regulierung der Akt-Aktivitäten sowie der anderen, der PI3K nachgeschalteten Signalkomponenten beteiligt ist.”
Und ich sag es noch mal: Signaltransduktionen sind keine Einbahnstraße.

“Wir berichten, dass cAMP die Proliferation sowohl über von der Proteinkinase A (PKA) abhängige als auch über PKA-unabhängige Signalwege stimuliert und dass die Phosphatidylinositol-3-Kinase (PI3K) für die cAMP-stimulierte Mitogenese erforderlich ist.”

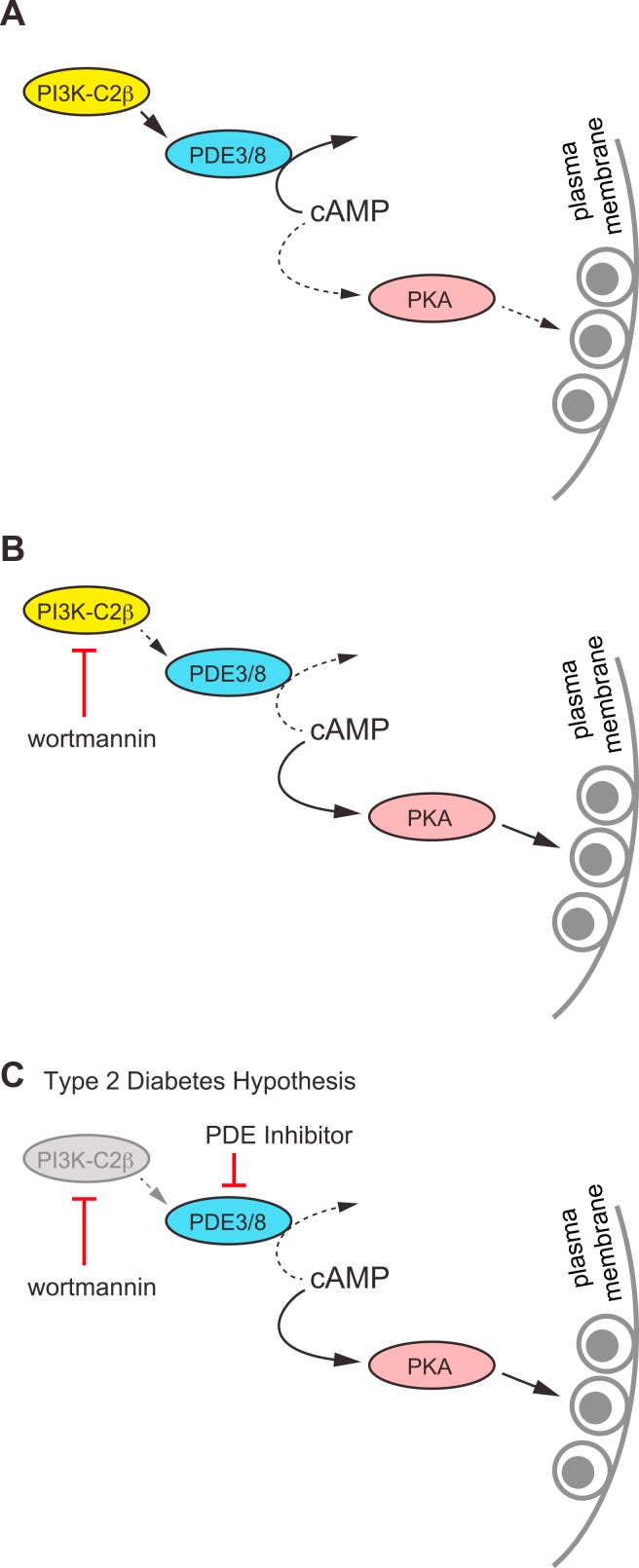
Uff! Damit haben wir also schon mal die erste von 4 der größten Signaltransduktionskaskaden halbwegs durchgeackert (keine Panik, wer sich mal mit der RHOA-Kaskade auseinandersetzt, weiß, dass diese gesamte Substackreihe nur als Lernprinzip dienen kann, was Crosstalks angeht). Wie ihr vielleicht bereits aus dieser einzigen Signalkaskade ableiten könnt, wäre es purer Wahnsinn außer Kontext Alterationen in der Aktivierung zu triggern, da ja das gleiche Basisprinzip für die anderen 3 Kaskaden gilt und somit ein nicht mehr überschaubares Netzwerk entsteht. Und es spielen ja auch noch extrazelluläre Faktoren, wie Zellmilieu (Zustand des extrazellulären Raumes um die Zelle), interzelluläre Crosstalks (Zelle zu Zelle Kommunikation via Exosome, Tight Junctions, elektrische Signale, Zytokine, usw. beispielsweise) und extrazelluläre Kommunikation (längere Strecken zu anderen Organen und darin befindlichen Zellen. Beispiel: Gehirn zu Darm oder Darm zu Gehirn → elektrische Signale, Zytokine, Hormone, etc, etc, etc.) eine essentielle Rolle und all das wird exakt durch diese Signale reguliert und gesteuert und steuert diese zeitgleich. Und wie aus all diesen Papers unschwer erkennbar sein dürfte, ist jede Interaktion absolut kontextspezifisch. Und exakt da ist auch der Hund begraben: Wir verstehen viele dieser Kontexte noch nicht.
Doch weiter im Programm.
JAK/STAT
cAMP auf JAK/STAT hatten wir ja schon.
Hier also umgedreht: JAK2 stimuliert direkt einen Teil von cAMP-Kaskade:

“PRL löst die JAK2/STAT5-abhängige Transkription in Nebennierenzellen aus, was jedoch keinen Einfluss auf die Corticosteron-Freisetzung hat. Im Gegensatz dazu offenbart die pharmakologische oder siRNA-vermittelte Hemmung von JAK2 dessen wesentliche Rolle sowohl bei der basalen als auch bei der ACTH/cAMP-induzierten Steroidogenese.”
JAK/STAT und die MAPks (mit extra vielen k):

“Die Hemmung der IL-6-Signalübertragung durch IL-1 und Proteinsynthese-Inhibitoren hing von der p38-Stresskinase ab, und die Aktivierung von p38 als Folge der induzierbaren Expression von MKK6 war ausreichend, um die IL-6-Signalübertragung zu hemmen.”
“Eine Rolle von p38 bei der Vermittlung der Hemmung legt nahe, dass mehrere Zytokine und Stressfaktoren, die p38-Signalwege in Monozyten aktivieren, wie IL-1, TNF, Toll-like-Rezeptoren und Fc-Rezeptoren, die Jak-STAT-Signalgebung durch pleiotrope Zytokine wie IL-6 modulieren können.”


“Die Behandlung der Zellen mit dem MEK-1-Inhibitor PD98059 führte zu einer signifikanten Verringerung der Phosphorylierung von STAT-1-alpha und IkappaB, was auf eine Wechselwirkung zwischen den ERK-1/2- und STAT-1-Signalwegen schließen lässt. Darüber hinaus führte die Exposition normaler menschlicher Bronchialepithelzellen gegenüber PD98059 (MEK-1-Inhibitor), AG490 (JAK-2-Inhibitor) oder Ro318220 (PKC-Inhibitor) zu einer signifikanten Verringerung der Anzahl infizierter Zellen, was darauf schließen lässt, dass ERK-1/2, JAK-2 und PKC für eine erfolgreiche RSV-Infektion in Bronchialepithelzellen erforderlich sind.
Schlussfolgerungen
Diese Ergebnisse zeigen, dass die ERK-1/2-Aktivierung für die RSV-induzierte frühe Genexpression erforderlich ist und dass der Cross-Talk zwischen ERK-1/2 und STAT-1 für die RSV-induzierte Expression proinflammatorischer Moleküle und die mögliche Verschlimmerung von Asthma entscheidend ist.”

“Diese Ergebnisse deuten darauf hin, dass die MAPK-Signalisierung und die STAT-Aktivierung sowohl bei Mäusen als auch bei menschlichen Brusttumoren in einem umgekehrten Verhältnis zueinander stehen. Diese Arbeit unterstützt auch die weitere Untersuchung von MEKi, um die STAT-Signalisierung und möglicherweise die Immuntherapie durch eine erhöhte MHC-I- und PD-L1-Expression zu verstärken.”
Bedenkt hierbei, dass ihr jeden Tag tausende entartete Zellen normalerweise durch intrazelluläre und immunzellgetriebene (IMHO falsch als adaptive und native betitelt) Immunprogramme, die ebenfalls durch diese Signalkaskaden angesteuert und reguliert werden, in Schach haltet.
Und wie immer: Es ist keine Einbahnstraße:
Blocking STAT3 signaling augments MEK/ERK inhibitor efficacy in esophageal squamous cell carcinoma (2022)

“Inhibitoren der Mitogen-aktivierten Proteinkinase-Kinase (MEK) blockierten effizient die Phosphorylierung der extrazellulären signalregulierten Kinase 1/2 (ERK1/2) bei ESCC, während die Signaltransduktion und der Aktivator der Transkription 3 (STAT3) rasch aktiviert wurden. Die kombinierte Hemmung von STAT3 verhinderte die Entstehung von Resistenzen und verstärkte den durch MEK-Inhibitoren induzierten Zellzyklusstillstand und die Seneszenz in vitro und in vivo. Mechanistische Untersuchungen ergaben, dass der Suppressor of Cytokine Signaling 3 (SOCS3) herunterreguliert wurde, was zu einem Anstieg der STAT3-Phosphorylierung in MEK-inhibierten Zellen führte. Darüber hinaus zeigte die Chromatin-Immunpräzipitation, dass ELK1, das durch MEK/ERK-Signale aktiviert wurde, die SOCS3-Transkription induzierte. Diese Daten deuten darauf hin, dass die Entwicklung einer kombinierten MEK- und STAT3-Inhibition eine nützliche Strategie für eine gezielte ESCC-Therapie sein könnte.”
Ich find es so mordswitzig, wie schon wieder die Hybris durchschlägt und man von einer nicht wirklich gut charakterisierten und verstandenen Interaktion, die man gerade mal anfängt zu beobachten, direkt auf therapeutische Ansätze schlussfolgern will (Cash cow. MUH!)
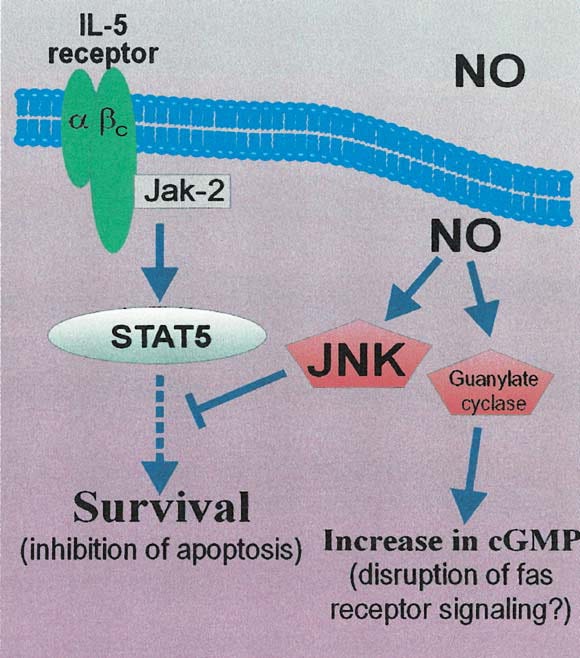
Ist es nicht faszinierend, wie komplex dieses Netzwerk ist? Hüpfen wir noch fix durch die Crosstalks zu mTORC1/2?
Dieses wirklich supergeile Paper, macht einfach nur Spaß:
The JAK/STAT signaling pathway: from bench to clinic


“Hier berichten wir, dass die mTORC2-Funktion zur Stabilisierung der p38-MAPK-Phosphatase DUSP10 führt und dadurch die p38-Aktivität hemmt.”
The mTORC2 signaling network: targets and cross-talks

Die PI3K/AKT-Crosstalks der mTOR-Komplexe haben wir ja schon beim PtdIns-Zyklus diskutiert gehabt[siehe Teil 3]. Also werde ich mir das jetzt mal schenken und noch auf ein weiteres, oftmals unterschätztes, grundlegendes Problem aufmerksam machen, was wir generell bei Beobachtungen für intrazelluläre Interaktionen haben:
The 2022 Report on the Human Proteome from the HUPO Human Proteome Project
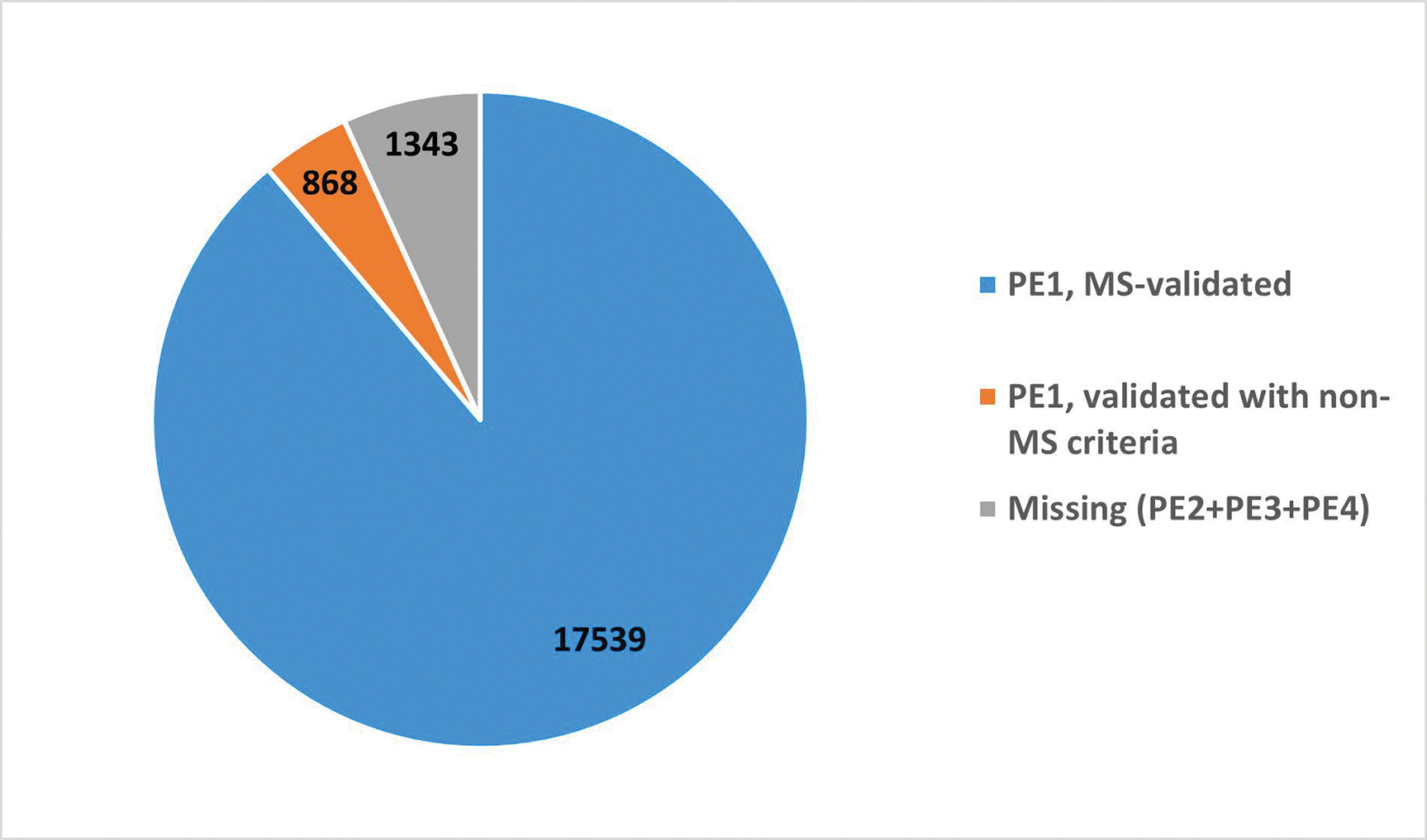
"Die "Metrics of the Human Proteome 2022" des HUPO Human Proteome Project (HPP) zeigen, dass die Proteinexpression nun für 18 407 (93,2 %) der 19 750 vorhergesagten Proteine, die im menschlichen Genom kodiert sind, glaubwürdig nachgewiesen wurde (neXtProt PE1-Level). Dies ist ein Nettozuwachs von 50 Proteinen seit 2021 aus Datensätzen, die weltweit generiert und vom HPP neu analysiert wurden. Umgekehrt hat sich die Zahl der fehlenden neXtProt PE2-, PE3- und PE4-Proteine um 78 von 1421 auf 1343 verringert. Dies bedeutet einen kontinuierlichen experimentellen Fortschritt bei der Liste der Proteomteile über alle Chromosomen hinweg sowie eine erhebliche Neuklassifizierung. Die Anwendung der Proteomik in einer Vielzahl von biologischen und klinischen Studien führt weiterhin zu bedeutenden Erkenntnissen und einer zunehmenden Integration mit anderen Omics-Plattformen. Wir stellen die Höhepunkte der Chromosomen-zentrierten HPP, der Biologie- und Krankheits-gesteuerten HPP und der HPP-Ressourcen-Säulen vor, vergleichen die Merkmale der Massenspektrometrie und der Olink- und Somalogic-Plattformen, stellen die Entstehung von Translationsprodukten aus der Ribosomen-Profilierung kleiner offener Leserahmen fest und diskutieren den Start des ersten HPP-Grand-Challenge-Projekts "A Function for Each Protein".”
Sind doch nur 2211 Proteine. 🤣
Wie viele Signalmoleküle werden wohl darunter sein? Macht nichts: Wir haben’s ja so perfekt begriffen, dass wir direkt einen nicht körpereigenen modRNA-Code mit völlig fremder Zusammensetzung und 3D-Struktur in unsere Zellen ballern konnten. Und dabei die Downstreamkaskaden, die zwingend durch die gesamte Umstrukturierung der Membran zu ignorieren, wird schon keine fatalen Konsequenzen nach sich ziehen. Prost Mahlzeit!
Ich spekuliere darauf, dass ich noch zwei weitere Teile in diesem Monstrum von Substack publizieren werde. Stay on tuned. Es wird nicht einfacher. 😜